Charge density wave and spin $1/2$ insulating state in single layer 1T-NbS$_2$
Cesare Tresca, Matteo Calandra

TL;DR
This study uses first-principles calculations to reveal that single-layer 1T-NbS2 exhibits a charge density wave and a spin-1/2 magnetic insulating state, with potential for synthesis similar to 1T-NbSe2.
Contribution
It demonstrates the existence of a charge density wave and magnetic insulating state in single-layer 1T-NbS2, highlighting its structural stability and magnetic properties using advanced computational methods.
Findings
1T-NbS2 has a $\sqrt{13} imes\sqrt{13}$ charge density wave.
The material exhibits a spin-1/2 magnetic state with a 0.15 eV band gap.
Exchange interactions suggest ferromagnetic coupling between stars.
Abstract
In bulk samples and few layer flakes, the transition metal dichalcogenides NbS and NbSe assume the H polytype structure with trigonal prismatic coordination of the Nb atom. Recently, however, single and few layers of 1T-NbSe with octahedral coordination around the transition metal ion were synthesized. Motivated by these experiments and by using first-principles calculations, we investigate the structural, electronic and dynamical properties of single layer 1T-NbS. We find that single-layer 1T-NbS undergoes a star-of-David charge density wave. Within the generalized gradient approximation, the weak interaction between the stars leads to an ultraflat band at the Fermi level isolated from all other bands. The spin-polarized generalized gradient approximation stabilizes a total spin magnetic state with opening of a eV band gap and…
Click any figure to enlarge with its caption.
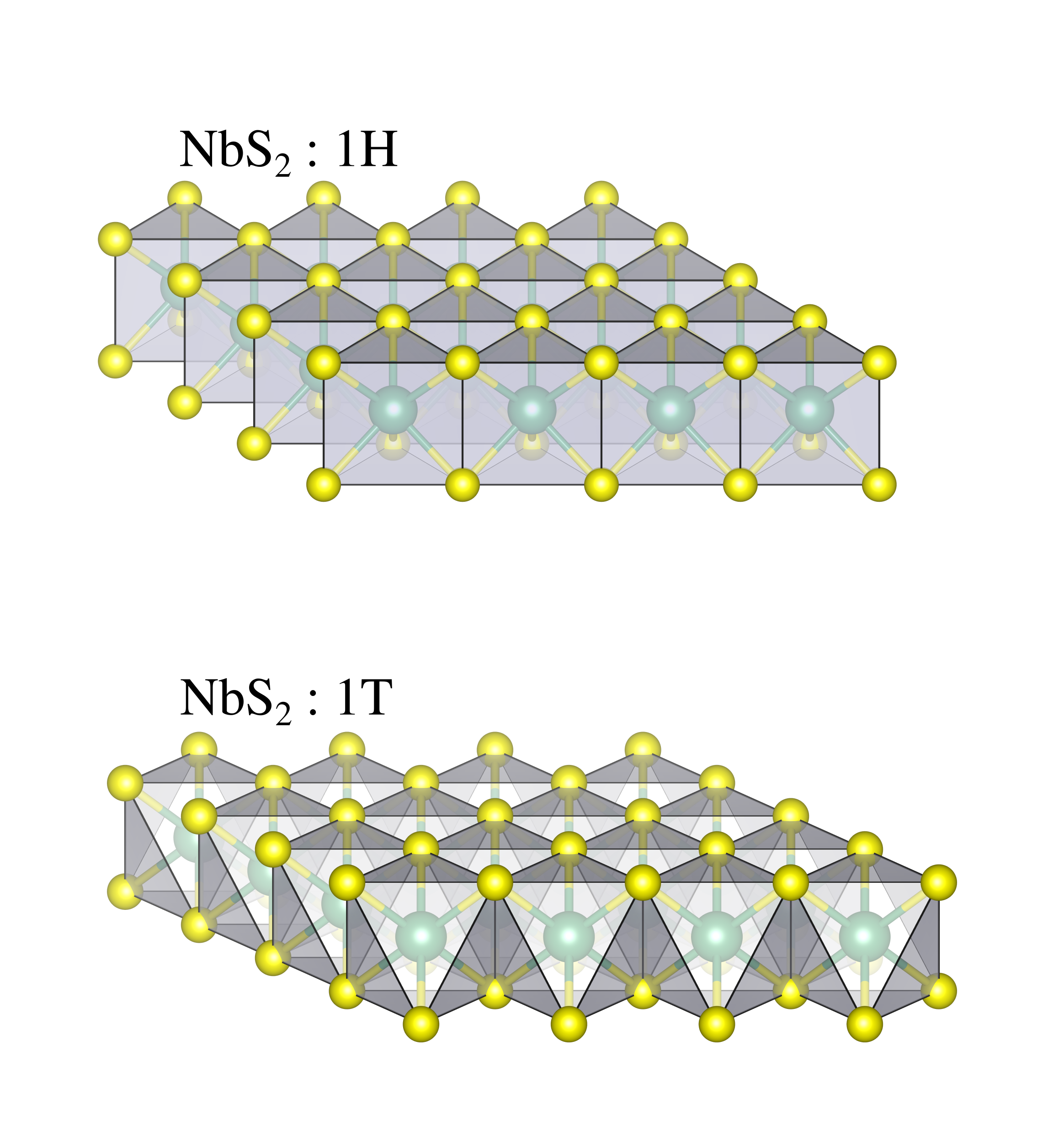 Figure 1
Figure 1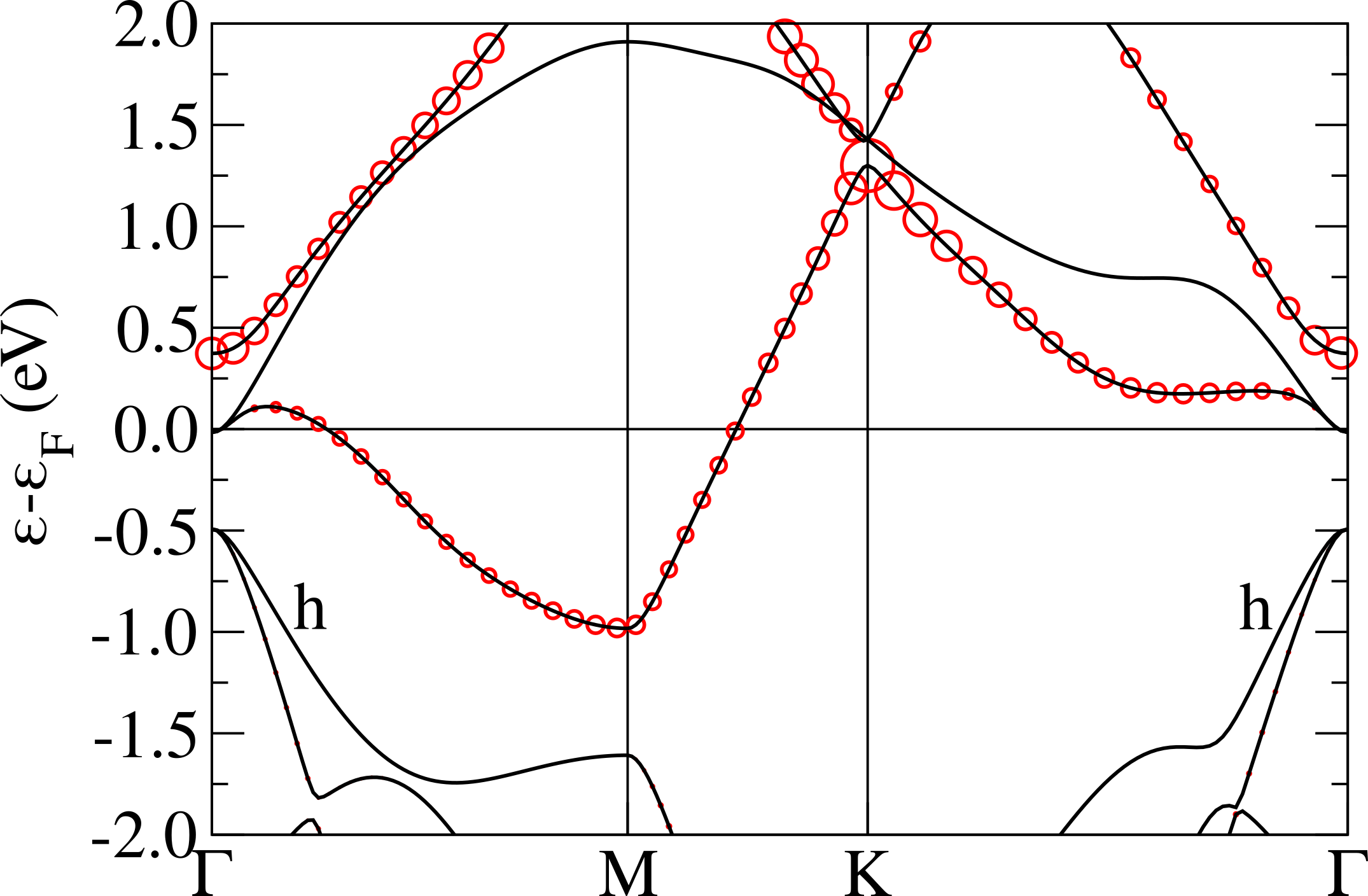 Figure 2
Figure 2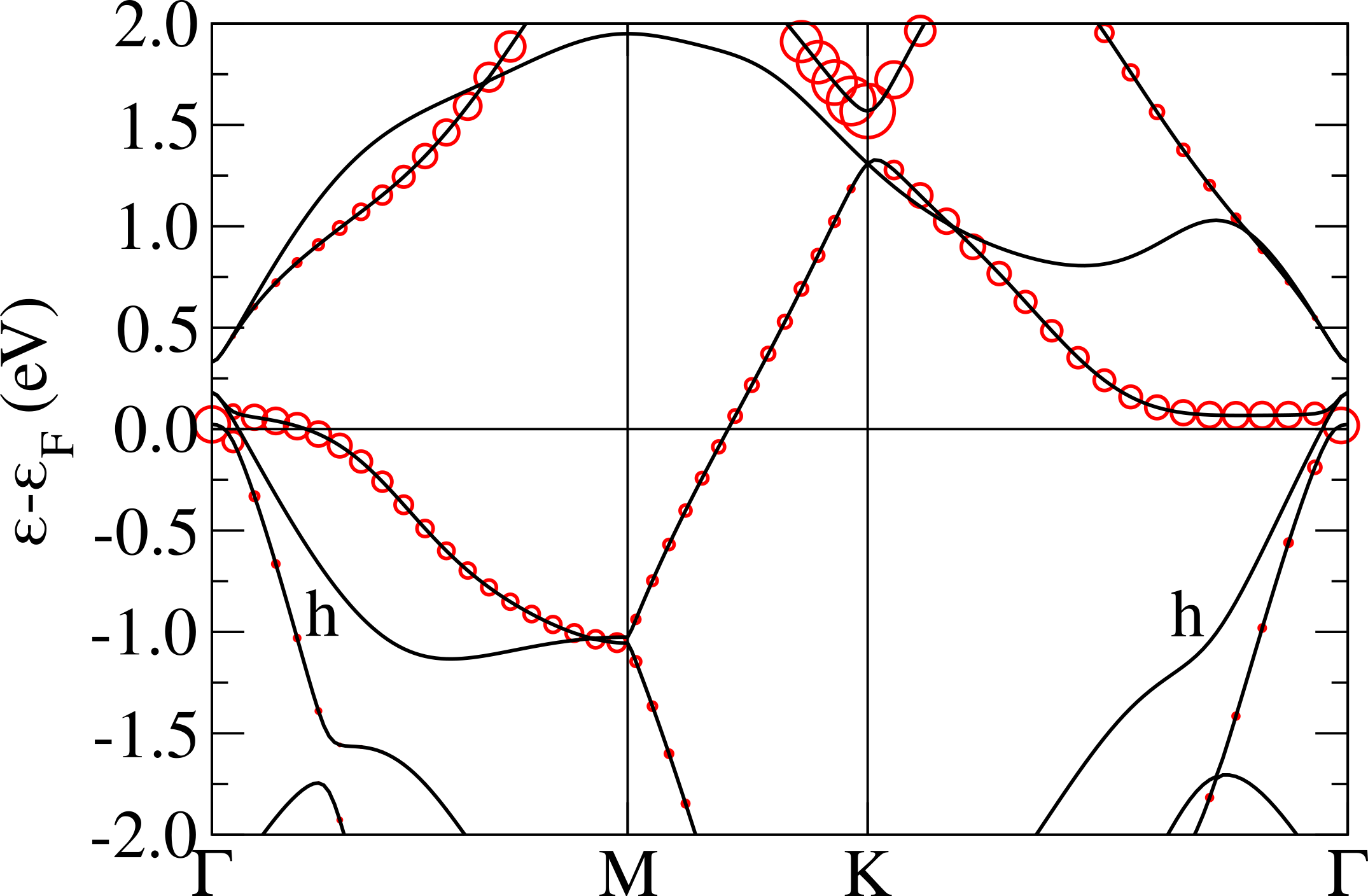 Figure 2
Figure 2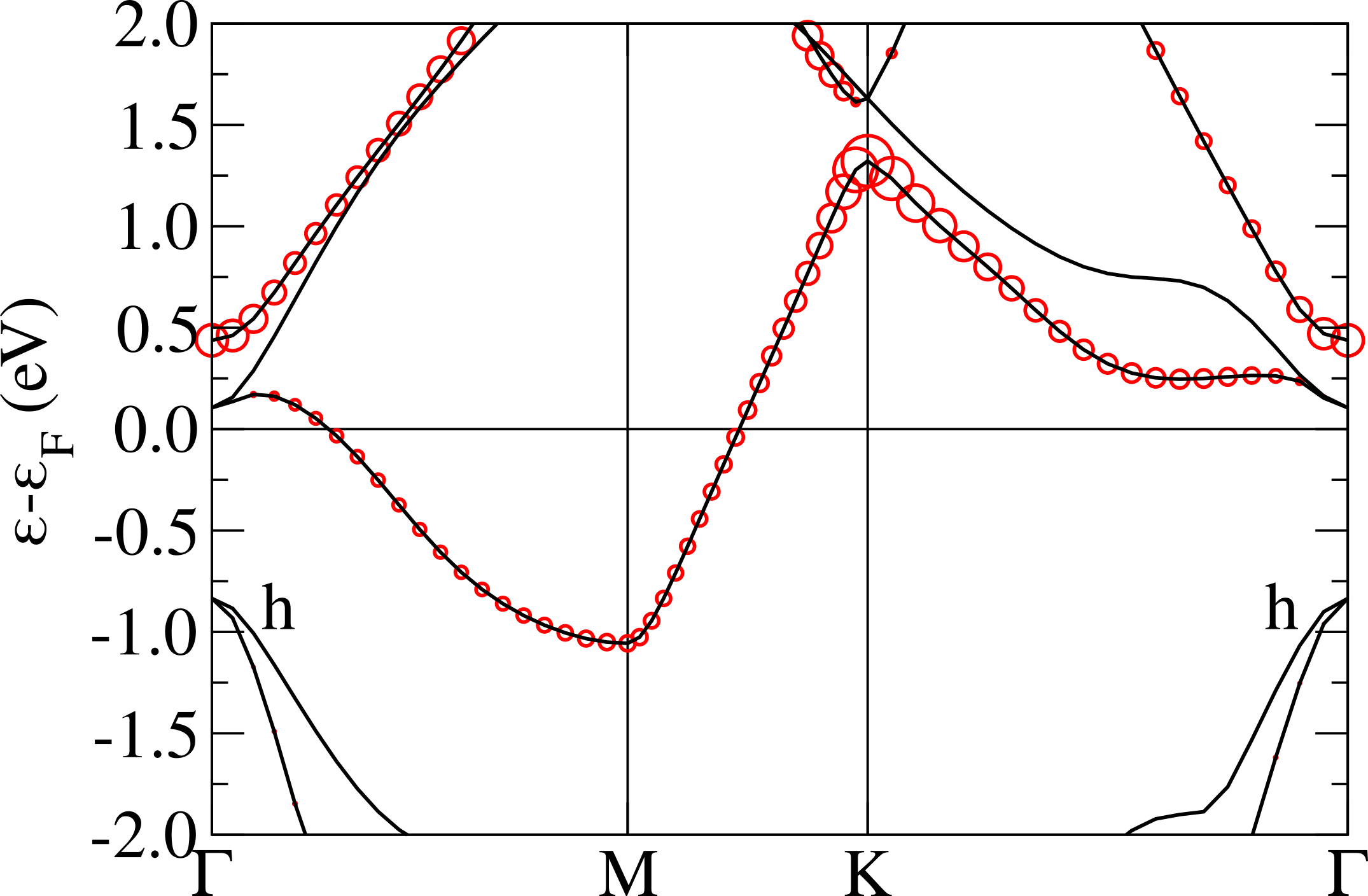 Figure 2
Figure 2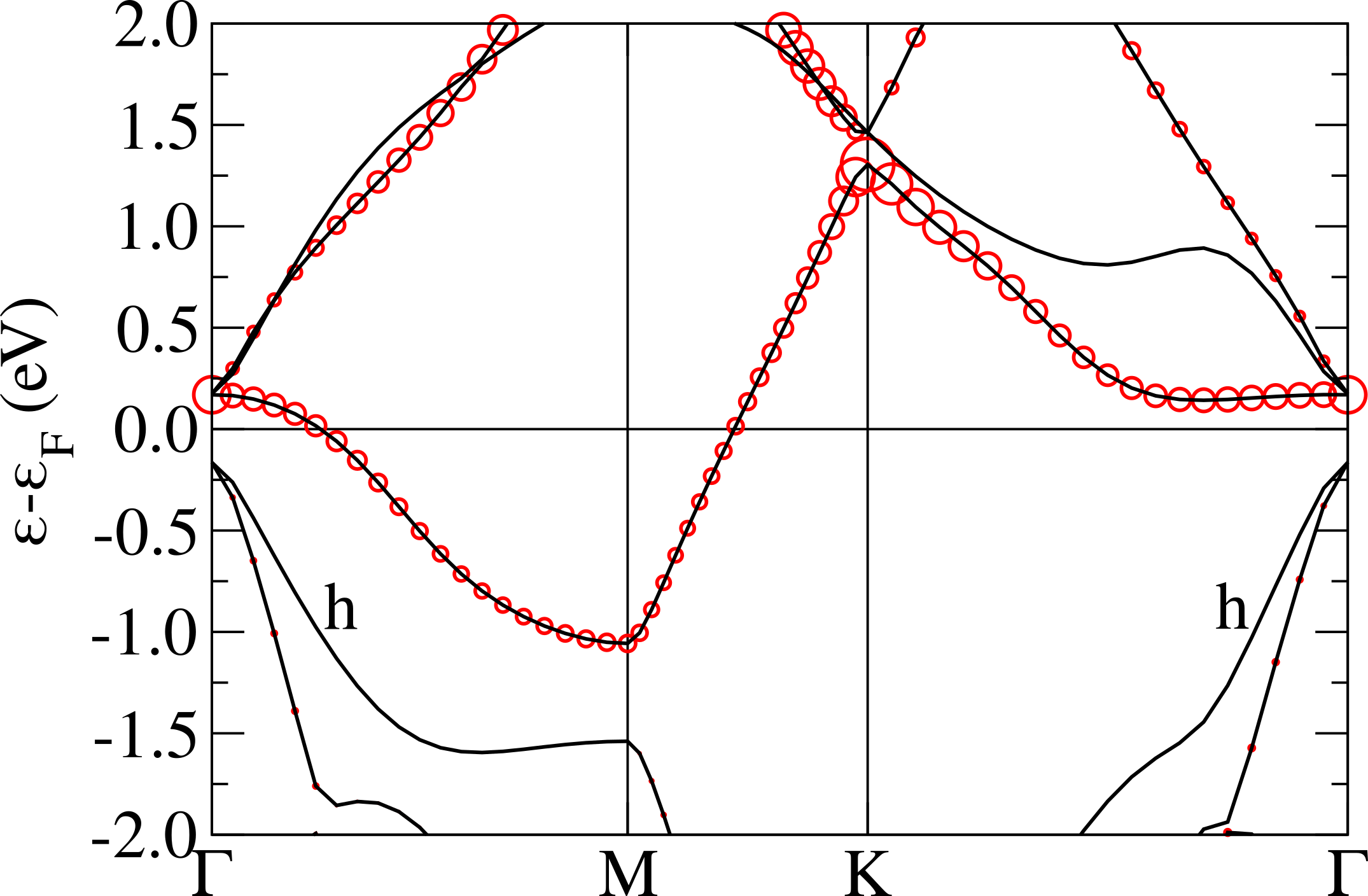 Figure 2
Figure 2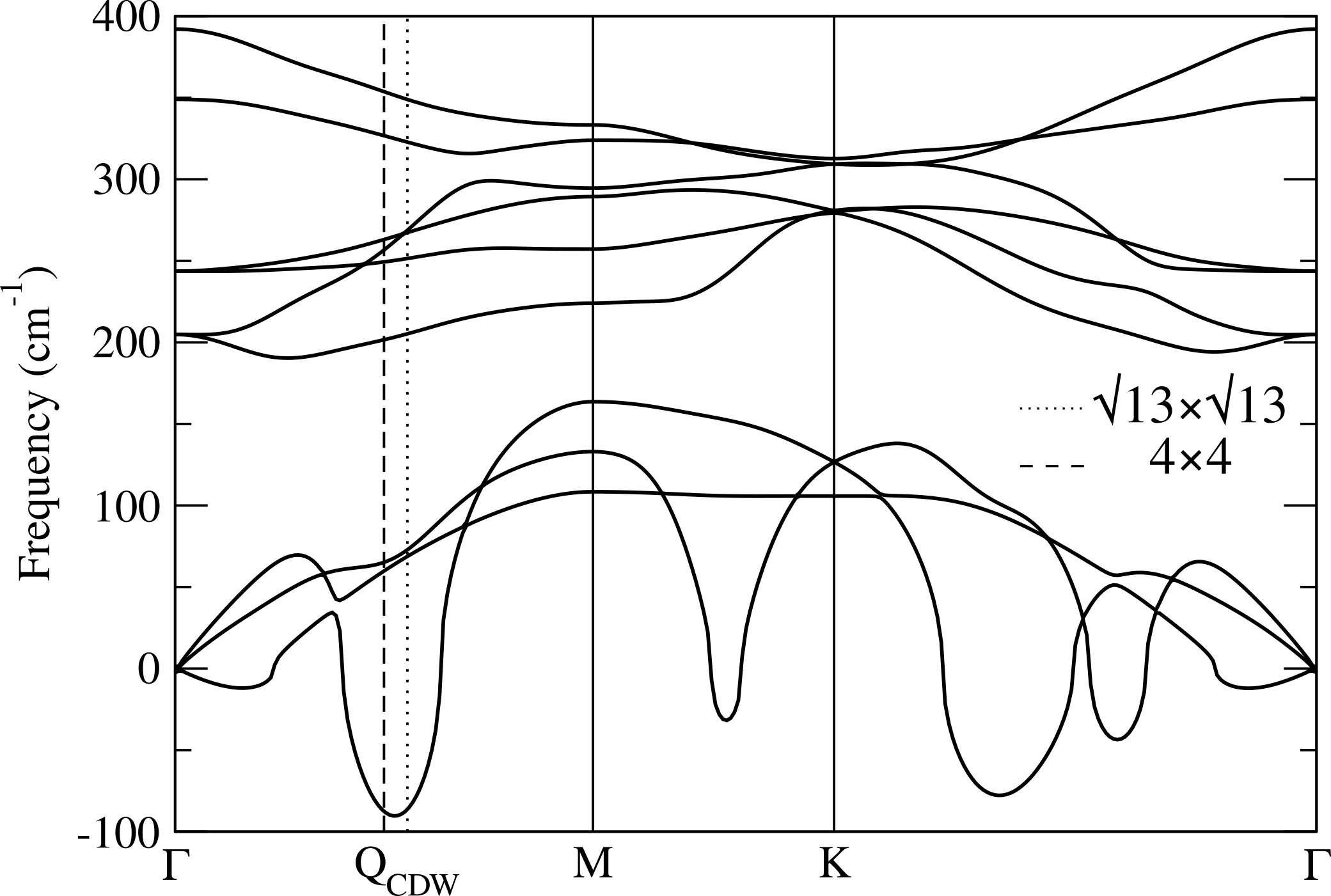 Figure 3
Figure 3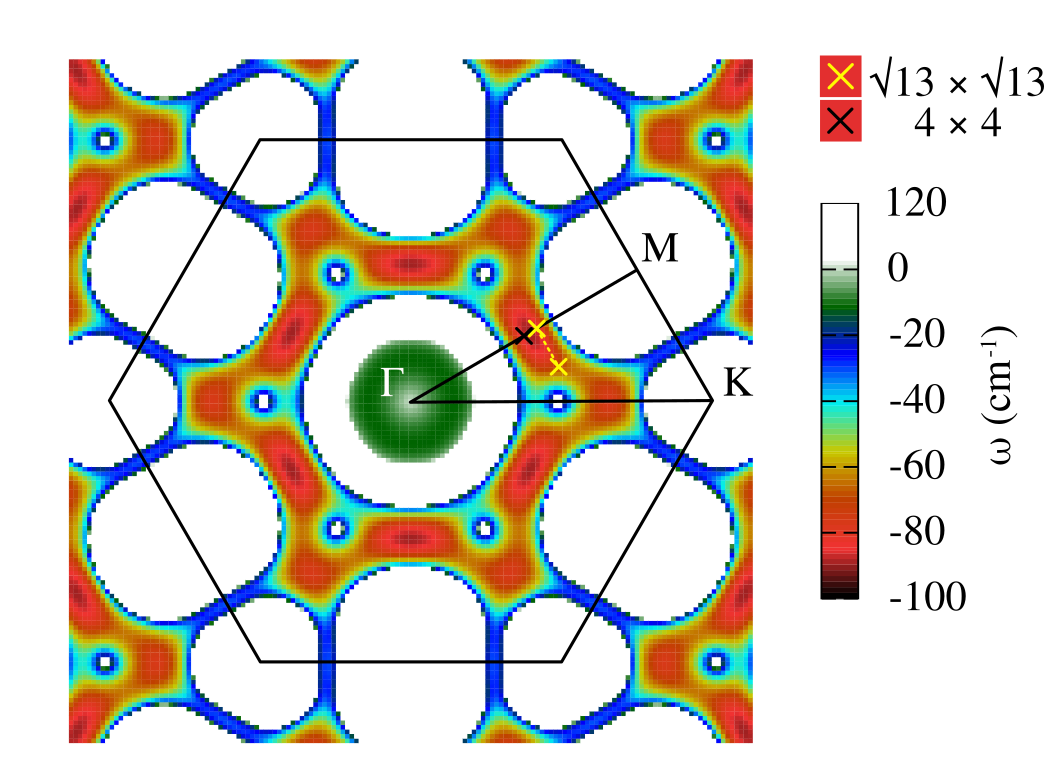 Figure 3
Figure 3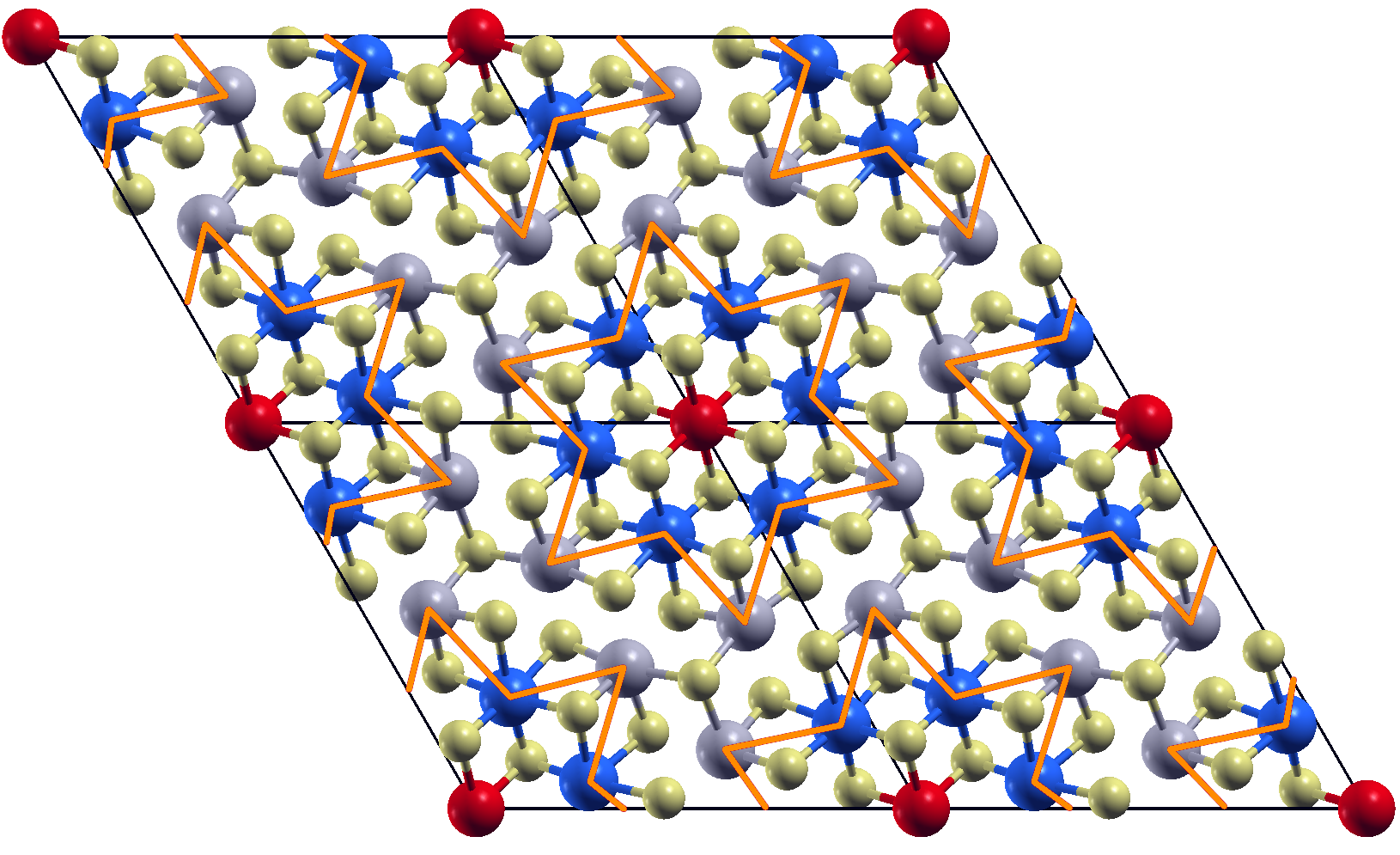 Figure 4
Figure 4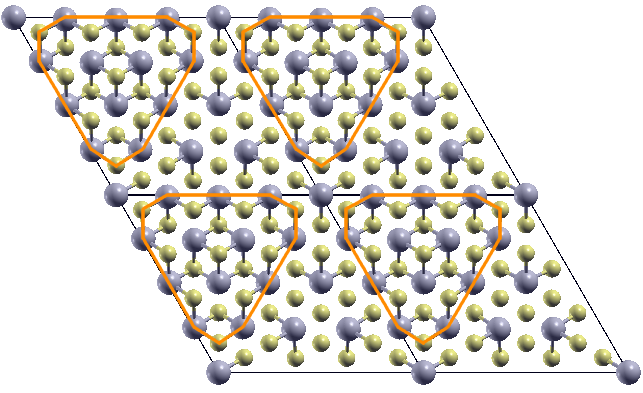 Figure 4
Figure 4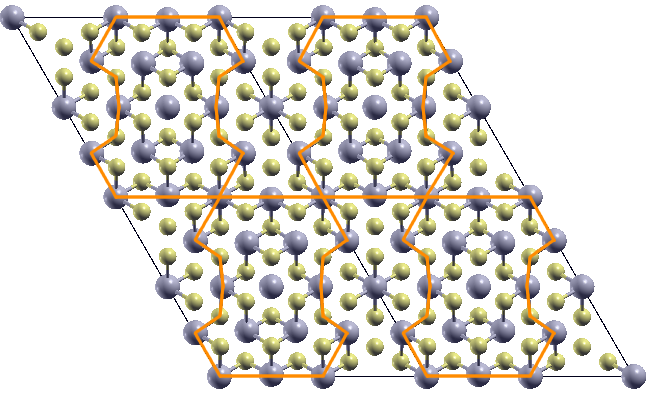 Figure 4
Figure 4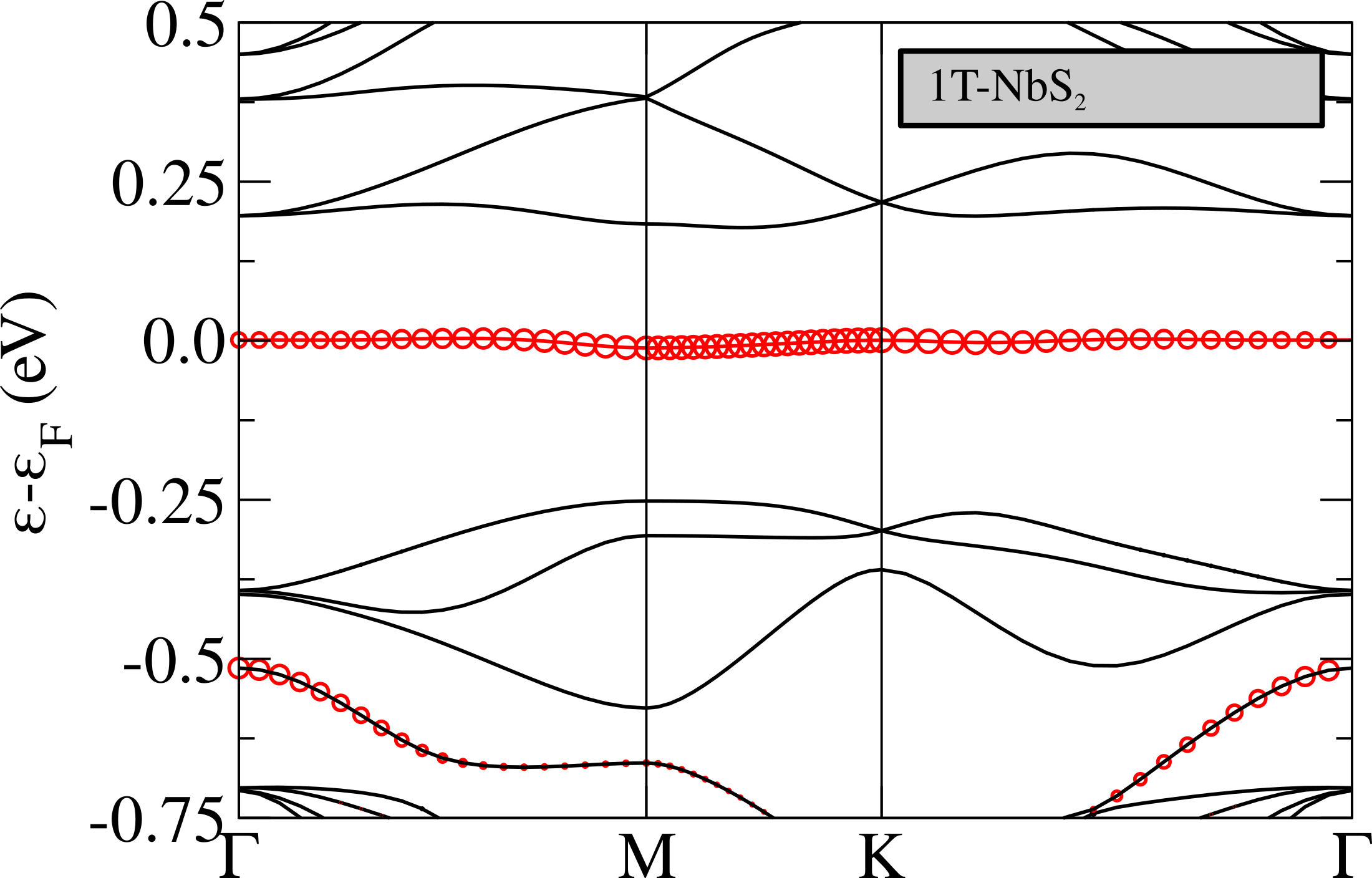 Figure 5
Figure 5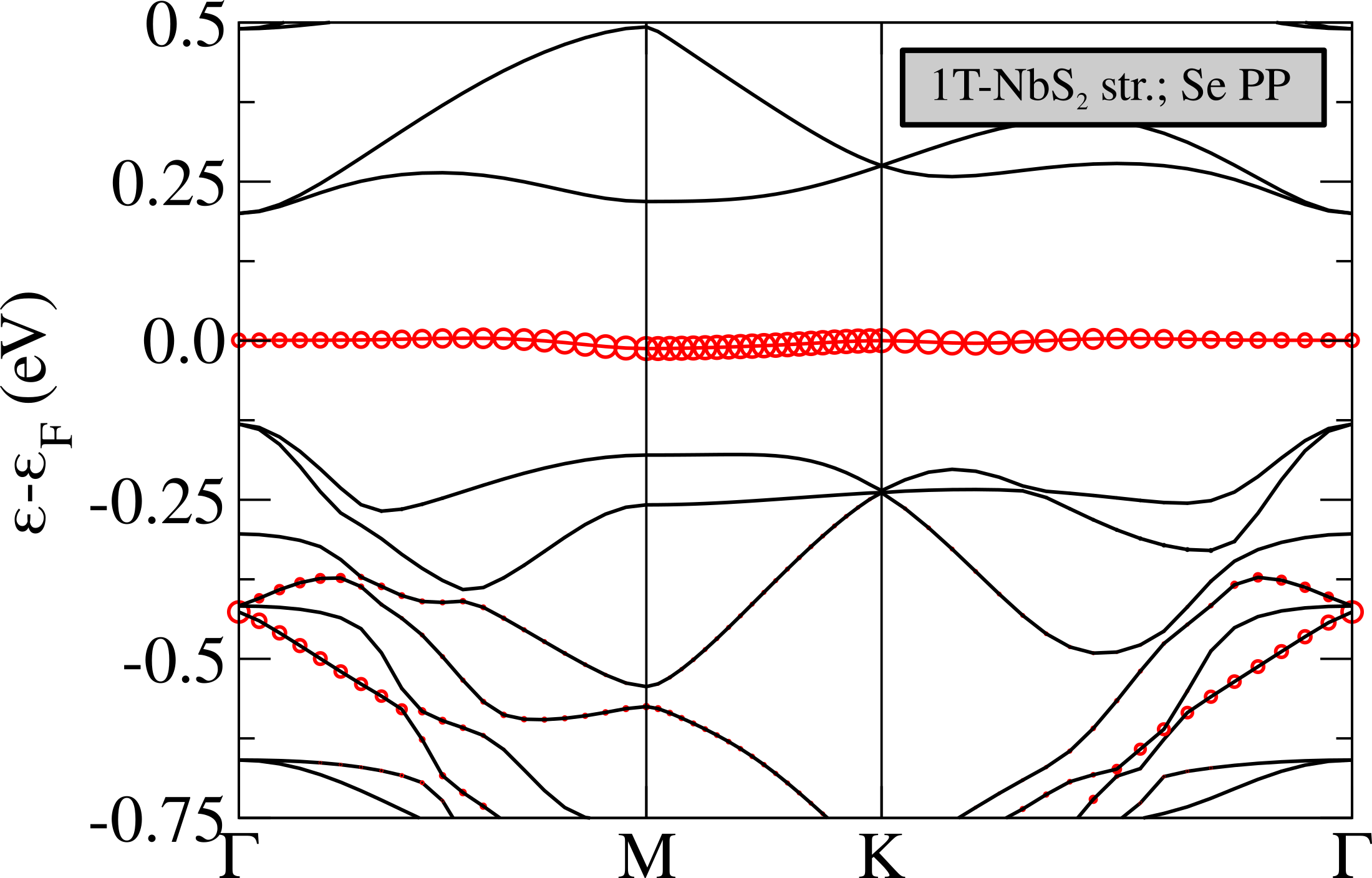 Figure 5
Figure 5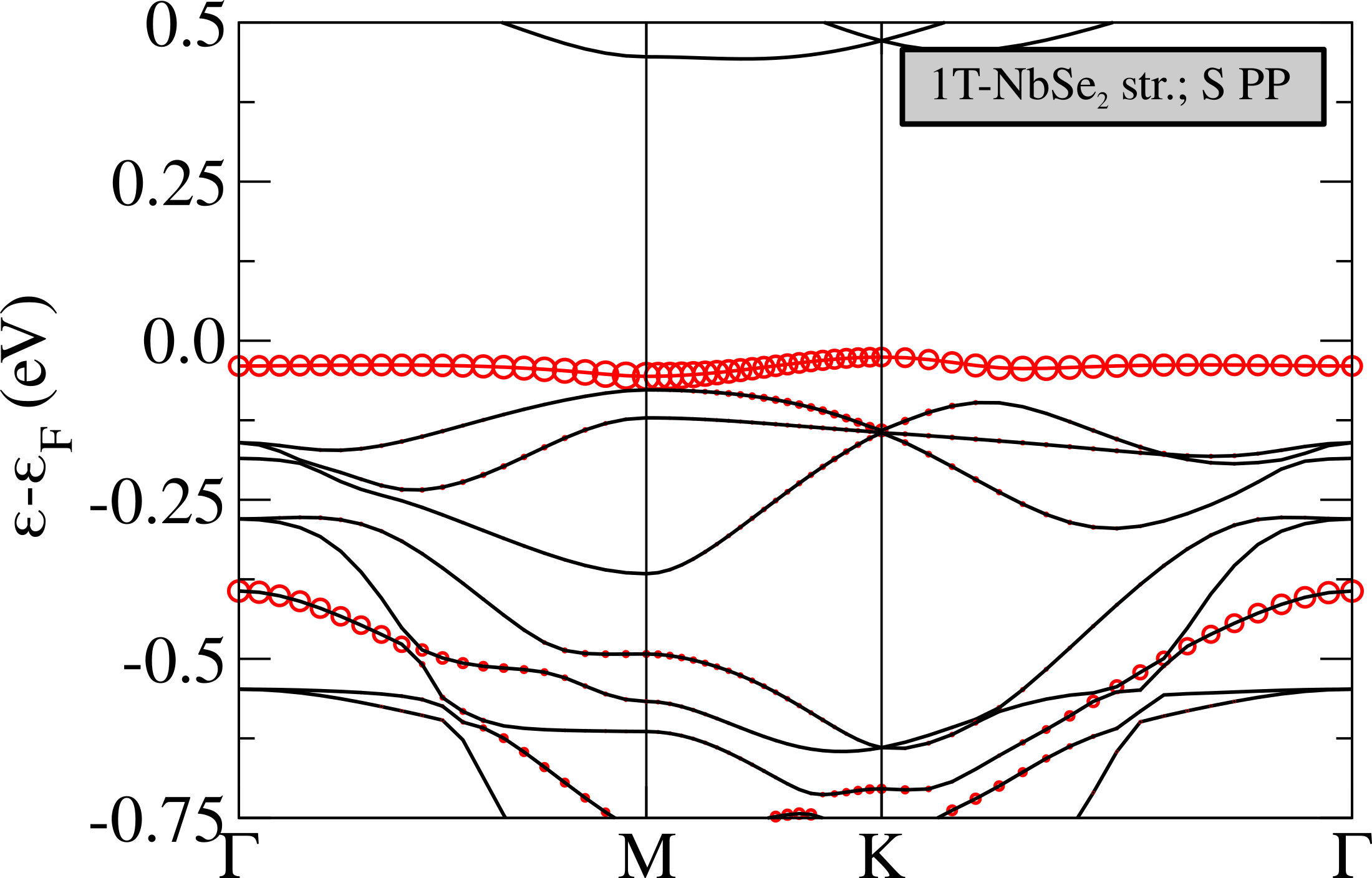 Figure 5
Figure 5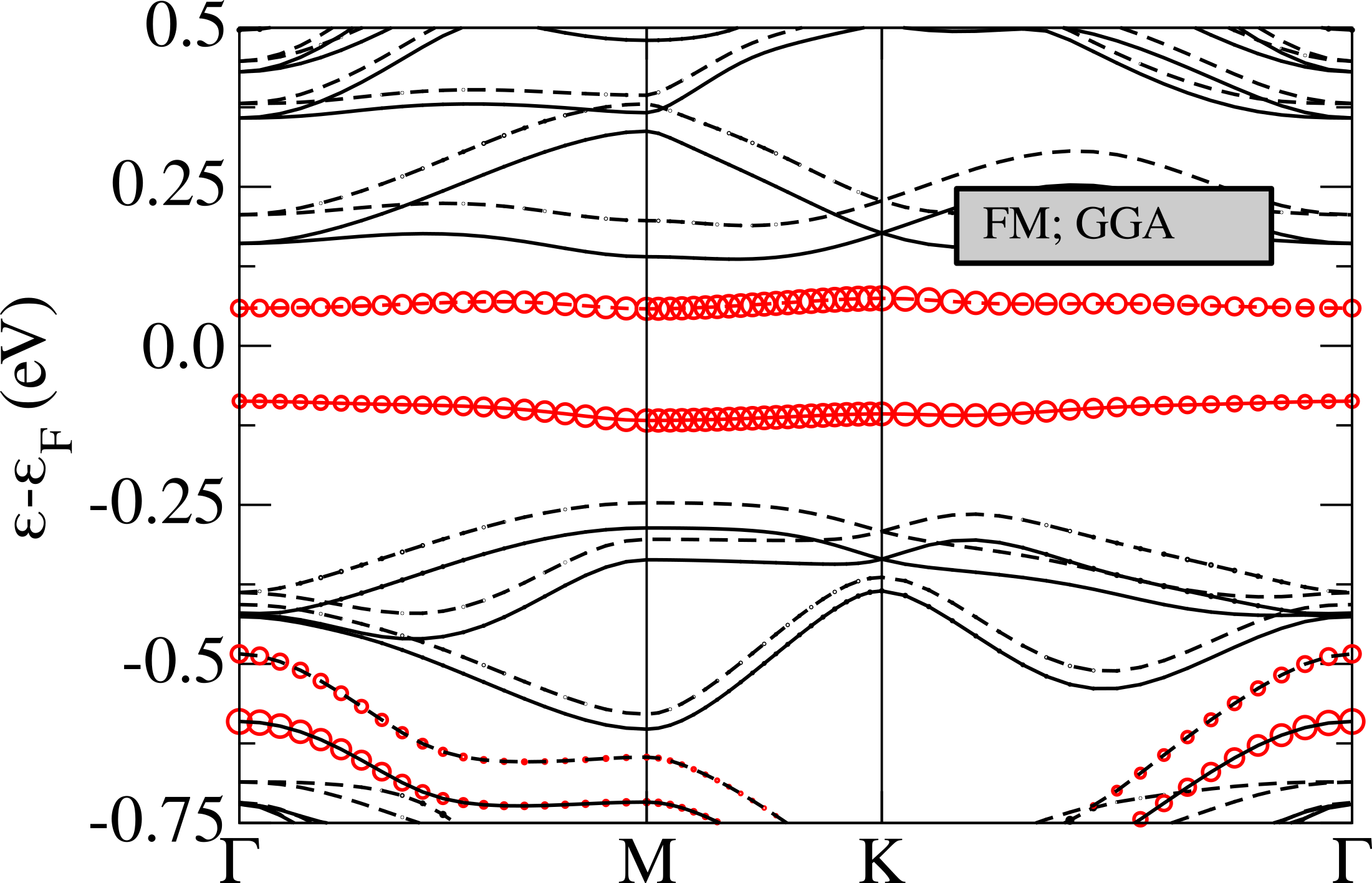 Figure 6
Figure 6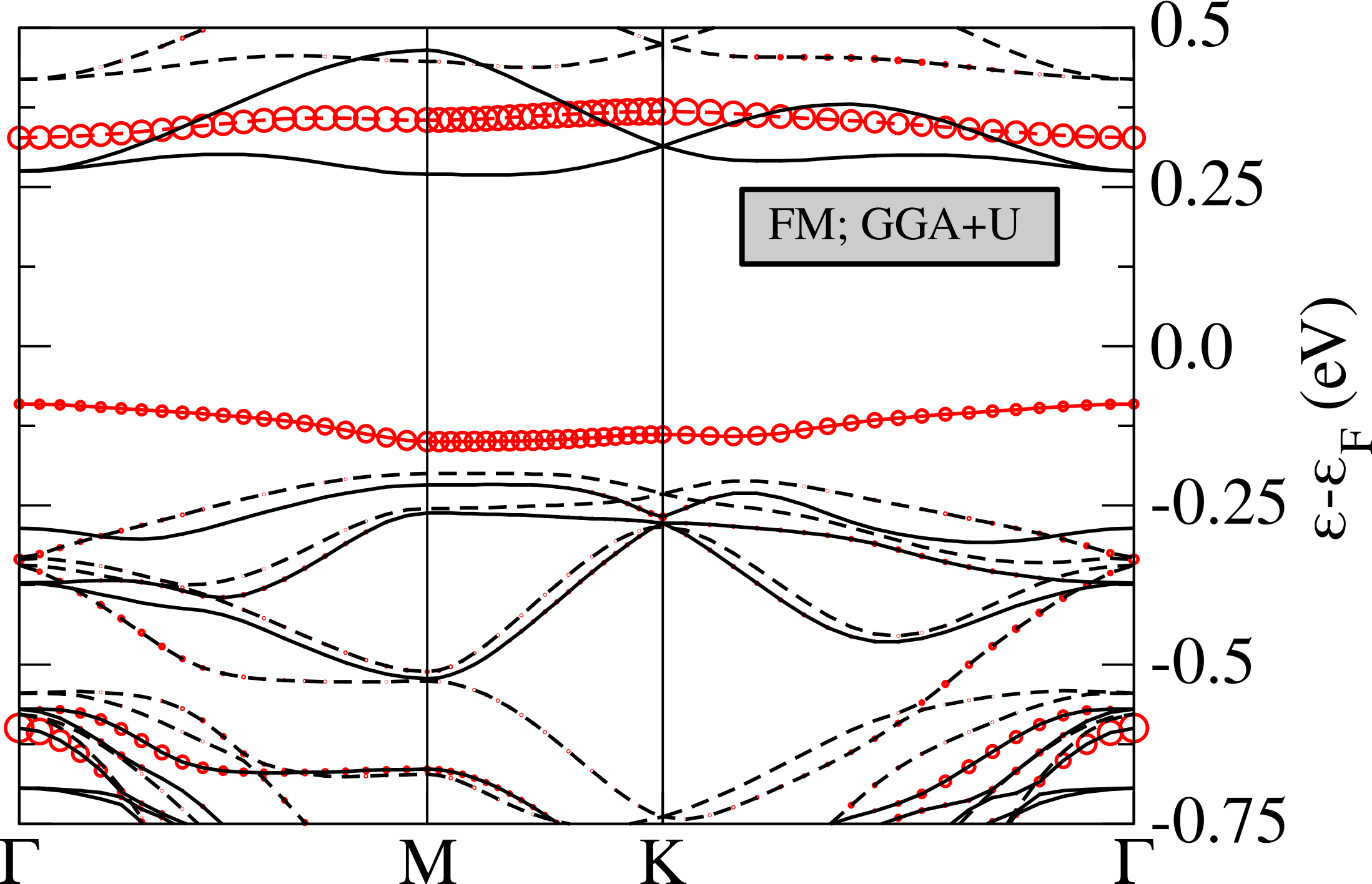 Figure 6
Figure 6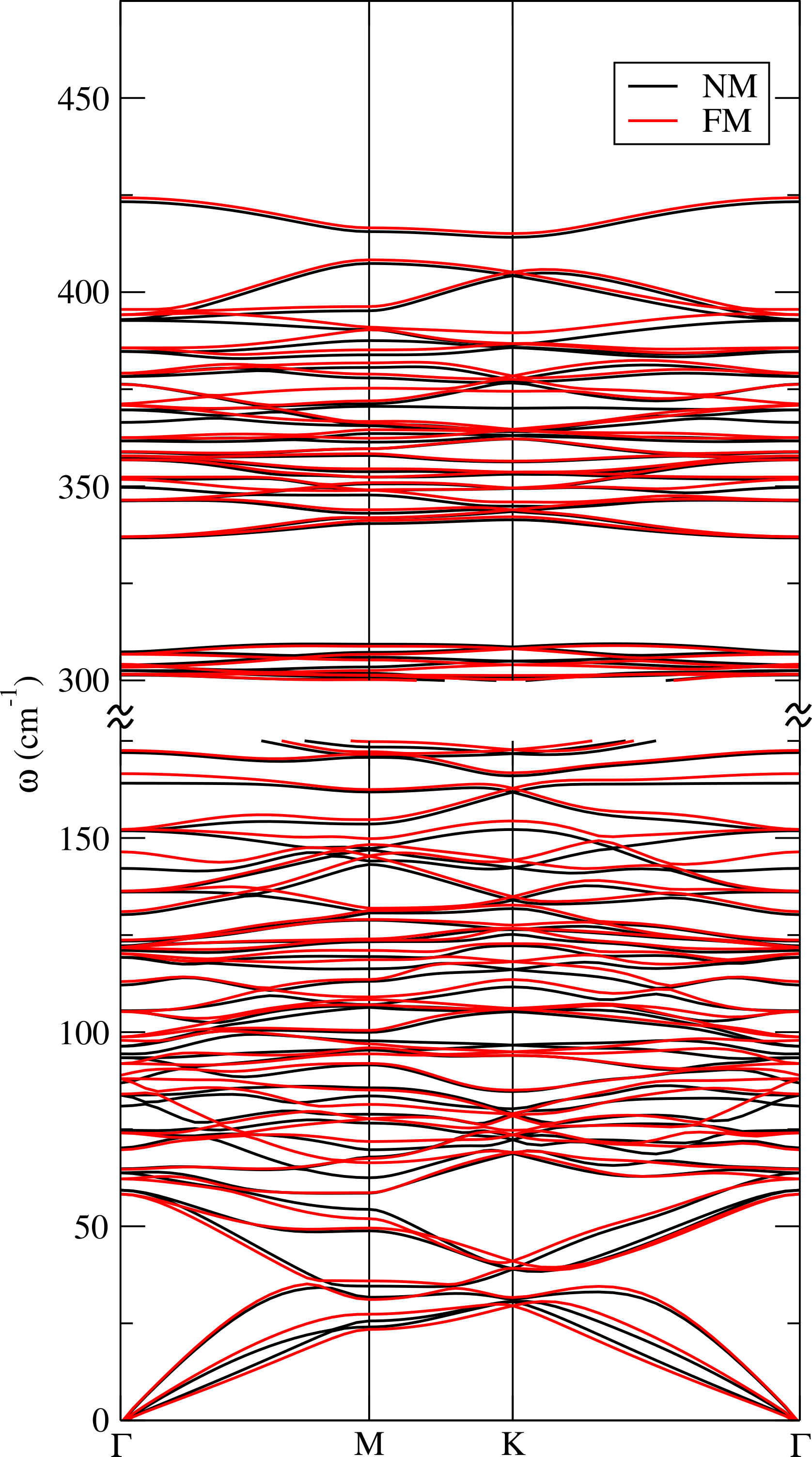 Figure 7
Figure 7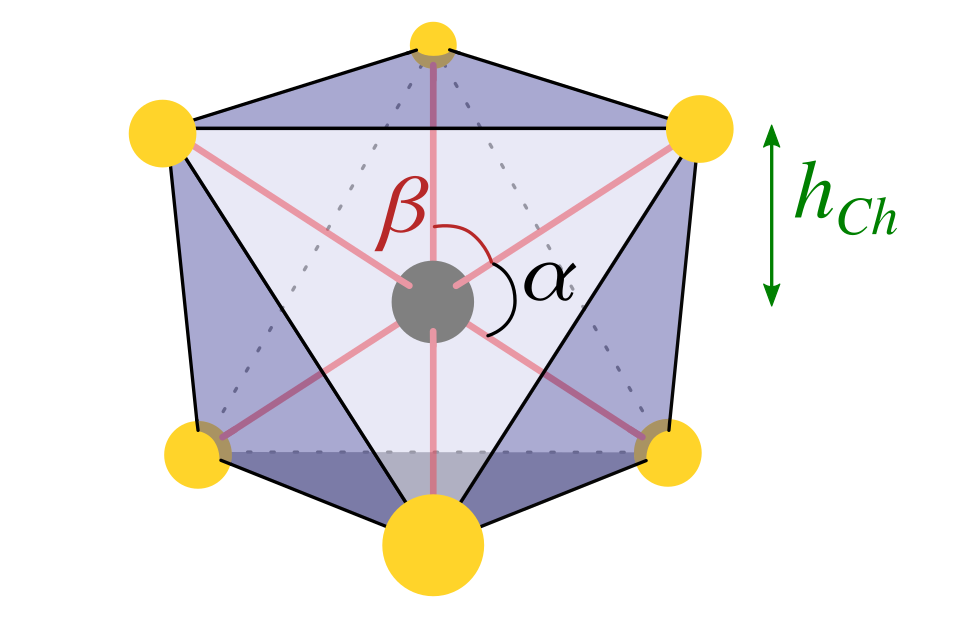 Figure 17
Figure 17| System | (Å) | (Å) | (deg) | (deg) | |
|---|---|---|---|---|---|
| NbS2 | 3.35 | 1.55 | 94.7 | 85.3 | |
| NbSe2 | 3.48Calandra (2018) | 1.69 | 97.5 | 82.5 | |
| TaS2 | 3.38 | 1.53 | 94.0 | 86.0 | |
| TaSe2 | 3.50 | 1.65 | 95.7 | 84.3 |
| System | ||||
|---|---|---|---|---|
| 1H | 0.0 | 0.0 | 3.346 | 3.326 |
| 1T | +7.2 | +4.2 | 3.360 | 3.357 |
| 44 | +5.8* | +1.9 | 13.485* | 13.428 |
| +4.3 | +1.2 | 12.200 | 12.123 | |
| (FM) | +4.4 | +0.5 | 12.198 | 12.126 |
| C=(0,0,0) | multiplicity | Wyckoff label | x | y | z |
|---|---|---|---|---|---|
| Nb | 1 | a | 0.00000 | 0.00000 | 0.00000 |
| Nb | 6 | g | 0.28854 | 0.07046 | 0.00044 |
| Nb | 6 | g | 0.63662 | 0.15249 | -0.00124 |
| S | 6 | g | 0.05083 | 0.17491 | 0.13591 |
| S | 6 | g | 0.35363 | 0.25132 | 0.13360 |
| S | 6 | g | 0.48561 | 0.19908 | 0.87839 |
| S | 2 | d | 0.33333 | 0.66667 | 0.88017 |
| S | 6 | g | -0.02692 | 0.40822 | 0.11972 |
| C=(0,0,0) | multiplicity | Wyckoff label | x | y | z |
|---|---|---|---|---|---|
| Nb | 1 | a | 0.00000 | 0.00000 | 0.00000 |
| Nb | 6 | g | 0.28891 | 0.07059 | 0.00036 |
| Nb | 6 | g | 0.63634 | 0.15237 | -0.00097 |
| S | 6 | g | 0.05072 | 0.17451 | 0.13662 |
| S | 6 | g | 0.35369 | 0.25129 | 0.13419 |
| S | 6 | g | 0.48561 | 0.19901 | 0.87758 |
| S | 2 | d | 0.33333 | 0.66667 | 0.87948 |
| S | 6 | g | -0.02695 | 0.40829 | 0.12062 |
| Point | ||
|---|---|---|
| group | (cm-1) | (cm-1) |
| Eg | 63.9 | 61.6 |
| Ag | 64.6 | 64.6 |
| Ag | 82.1 | 83.2 |
| Eg | 84.4 | 88.6 |
| Eg | 93.3 | 91.5 |
| Ag | 94.2 | 97.9 |
| Eg | 106.0 | 105.4 |
| Eg | 119.5 | 120.2 |
| Ag | 121.2 | 121.7 |
| Ag | 142.9 | 146.3 |
| Ag | 164.0 | 166.7 |
| Eg | 179.7 | 180.4 |
| Ag | 211.1 | 211.1 |
| Eg | 218.8 | 218.8 |
| Ag | 221.3 | 221.6 |
| Eg | 226.9 | 227.1 |
| Eg | 236.2 | 236.3 |
| Eg | 242.2 | 242.4 |
| Ag | 243.7 | 243.6 |
| Eg | 257.7 | 258.6 |
| Eg | 261.6 | 261.7 |
| Ag | 262.3 | 262.7 |
| Ag | 266.8 | 266.7 |
| Eg | 268.1 | 268.6 |
| Eg | 276.7 | 276.9 |
| Ag | 283.9 | 283.6 |
| Ag | 286.0 | 285.5 |
| Ag | 298.7 | 298.7 |
| Ag | 303.4 | 303.2 |
| Eg | 307.5 | 306.9 |
| Eg | 346.5 | 346.5 |
| Ag | 349.9 | 351.7 |
| Eg | 357.5 | 357.9 |
| Ag | 358.9 | 359.1 |
| Ag | 366.7 | 371.0 |
| Eg | 378.4 | 379.0 |
| Ag | 392.8 | 394.1 |
| Eg | 393.0 | 395.1 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Charge density wave and spin insulating state in
single layer 1T-NbS2.
Cesare Tresca
Matteo Calandra
Sorbonne Université, CNRS, Institut des Nanosciences de Paris, UMR7588, F-75252, Paris, France
Abstract
In bulk samples and few layer flakes, the transition metal dichalcogenides NbS2 and NbSe2 assume the H polytype structure with trigonal prismatic coordination of the Nb atom. Recently, however, single and few layers of 1T-NbSe2 with octahedral coordination around the transition metal ion were synthesized. Motivated by these experiments and by using first-principles calculations, we investigate the structural, electronic and dynamical properties of single layer 1T-NbS2. We find that single-layer 1T-NbS2 undergoes a star-of-David charge density wave. Within the generalized gradient approximation, the weak interaction between the stars leads to an ultraflat band at the Fermi level isolated from all other bands. The spin-polarized generalized gradient approximation stabilizes a total spin magnetic state with opening of a eV band gap and a magnetic moment localized on the central Nb in the star. Within GGA+U, the magnetic moment on the central Nb is enhanced to and a larger gap occurs. Most important, this approximation gives a small energy difference between the 1T and 1H polytypes (only mRy/Nb), suggesting that the 1T-polytype can be synthesized in a similar way as done for single layer 1T-NbSe2. Finally we compute first and second nearest neighbors magnetic inter-star exchange interactions finding =9.5 K and =0.4 K ferromagnetic coupling constants.
I Introduction
Bulk transition metal dichalcogenides (TMDs) of the form TCh2, where T is a transition metal and Ch is a chalcogen (Se, S, Te), are very versatile systems as their electronic and structural properties can be tuned not only by varying their chemical composition but also by synthesizing different polytypes having the same chemical formula. The variation of the local coordination of the transition metal ion in different polytypes of a given TMD leads to completely different physical propertiesWilson et al. (1975). For example, 1T-TaS2 with Ta in octahedral coordination, is a correlated system which ground state is still very debated (Mott insulator or correlated metal) FAZ (1980); Fazekas and Tosatti (1979); Darancet et al. (2014); Ngankeu et al. (2017) while 2H-TaS2, with Ta in trigonal prismatic coordination, is a metal (the 1T and 1H polytypes are reported in Fig.1). However, this tunability cannot be completely exploited as not all bulk TMDs can be synthesized in 1T and 2H polytypes either because the appropriate chemical and thermodynamical preparation conditions are unknown or because the energetic is unfavorable. This is the case of bulk 1T-TiSe2 that has never been synthesized in the 2H polytype or, vice versa, of bulk 2H-NbSe2 and 2H-NbS2 that crystallizes in the 2H polytype and not in the 1T one, although it has been reportedCarmalt et al. (2004) that 1T-NbS2 bulk can be synthesized under very special conditions.
Since different TCh2 planes are bounded together by the weak van der Waals interaction, it makes possible to isolate single layers of a large class of transition metal dichalcogenides Novoselov et al. (2005); et al. (2011) (a single layer here refers to a trilayer TCh2 unit). The exfoliation of bulk TMDs into bi-dimensional (2D) crystals, beside being interesting in itself as it allows to investigate a variety of phenomena in low dimension, paves the way to different synthesis techniques, unfitted for bulk systems but feasible in few layers flakes. An example is the phase transition between the hexagonal and monoclinic phases of monolayer MoTe2 achieved by electrostatic dopingWang et al. (2017) or the transition between the 2H and 1T and 1T*′* phases obtained by liquid exfoliationet al. (2011); Eda et al. (2011).
More recently it has been shown that the 1T-NbSe2 polytype can be stabilized either as a single layer on top of bilayer graphene kept at 500-590 ∘C during epitaxyNakata et al. (2016) or by applying a pulsed local field through the STM tip at the surface of bulk 2H-NbSe2Bischoff et al. (2017).
The physical properties of single layer 1T-NbSe2 turned out to be completely different from that of single layer 1H-NbSe2 as the former is a spin Mott-Jahn Teller insulator undergoing a charge density waveNakata et al. (2016); Calandra (2018), while the latter is a metal undergoing a charge density waveXi et al. (2015); Ugeda et al. (2016); Calandra et al. (2009). Most important, it has been recently shownCalandra (2018) that density functional theory calculations (DFT) with local LDA/GGA kernels do not explain the stabilization of the 1T-NbSe2 single layer phase with respect to the 2H one, as this transition occurs via a correlated mechanism involving vibrations and the stabilization of a magnetic state that can be addressed within the DFT+U approximation. Thus, given the broad perspectives offered by these new synthesis techniques, theoretical calculations can be used to spot new TMD phase that can be experimentally accessed and to describe their structural and electronic properties.
Bulk 2H-NbS2 is isoelectronic and isostructural to 2H-NbSe2, however it stands somewhat at odd with respect to other transition metal dichalcogenides as it displays no charge density wave (CDW) at low temperatureLeroux et al. (2012). On the contrary, when NbS2 single-layer is grown on Nitrogen-doped 6H-SiC(0001) terminated with single or bilayer graphene, it adopts the 1H-NbS2 polytype and STM images show a reconstructionLin et al. (2018), but if 1H-NbS2 is grown epitaxially on Au(111) no charge density wave is detectedStan et al. (2019). Given the different properties of NbS2 in the 2D limit, it is natural to investigate the possible stability of other polytypes and the formation of magnetic and charge density wave phases.
In this work, by using density functional theory calculations, we investigate the possible synthesis of single layer 1T-NbS2 together with its structural, vibrational and electronic properties. We study the stability with respect to the single layer phase and we calculate magnetic couplings.
II Computational details
Density functional theory calculations are performed using the Quantum-Espresso code Giannozzi et al. (2009, 2017). For Nb (Ta) we use ultra-soft pseudopotentials from Vanderbilt distributionsVanderbilt (1990) including semicore states and two projectors for and channels and valence configuration , , , (, , , ). For S (Se) we use norm-conserving pseudopotentials with empty -states in valence and the following valence configuration , , (, , ).
We use an energy cutoff up to 45 Ry (540 Ry for the charge density) for all the calculations. For the exchange correlation energy we take the generalized gradient approximation (GGA) and the GGA+U one.
The charge density integration over the Brillouin Zone (BZ) is performed using an uniform Monkhorst and Pack grid Monkhorst and Pack (1976) for the 1T and 2H-polytypes ( and for the and CDW phases respectively) and a Ry Gaussian smearing. For the total energy comparison among magnetic solutions of the reconstruction we reduce the smearing to Ry increasing the BZ grid to (for the evaluation of exchange constants we use super-cells: we scale the BZ sampling grid to assure the same density used in the other calculations). The surface is simulated by considering a supercell with about Å of vacuum along the c-axis between the periodic images. We use the theoretical in-plane lattice parameters and perform full structural optimization of the internal degrees of freedom. Phonon modes in the undistorted 1T-phase are calculated in linear response theoryGiannozzi et al. (2009, 2017) over 19 phonon wave-vector mesh in the irreducible BZ using an uniform reciprocal space mesh for sampling the electronic states.
III Results and discussion
III.1 High-symmetry 1T-NbS2 structure.
We start by performing geometrical optimization of the undistorted 1T-NbS2 structure (3 atoms/cell). For completeness and to achieve a better understanding of the transition metal/chalcogen hybridization, we also calculate the theoretical GGA structural parameters and electronic structures of 1T-NbSe2, 1T-TaS2 and 1T-TaSe2 high symmetry phases. We obtain the lattice parameter and internal coordinates reported in Tab. 1. As it can be seen the sulfur dichalcogenides are somewhat compressed in the basal plane with respect to Se dichalcogenides. The smaller in-plane parameter is accompanied by a smaller chalcogen height () and a smaller tetragonal distortion of the octahedral crystal symmetry around the transition metal ion.
This is relevant as the electronic structures of all these highly symmetric polytypes are similar but with important differences that can be in part attributed to the amount of Jahn-Teller trigonal distortion of the octahedral crystal field around the transition metal and in part to the alignment of the chalcogen and transition metal levelsWhangbo and Canadell (1992); Mattheiss (1973).
In more details, the octahedral crystal field splitting leads to triply degenerate t2g orbitals (, , ) and doubly degenerate eg orbitals (, ) at higher energy (we adopt here the same convention of Ref. Mattheiss (1973) for the crystal axes). The trigonal distortion of the octahedron is identified by the bond angles centered at the transition metal ion and having bonds to the nearest chalcogens (see picture in Tab. 1). In an undistorted octahedron , while in the present cases there is a substantial deviation from the ideal values.
The crystal field for a trigonal distorted octahedron splits the t2g orbitals in a twofold degenerate state (, ) and a single degenerate state. We label this energy separation at zone center “apparent Jahn-Teller splitting”. In the case of 1T-NbS2 and 1T-TaS2, the apparent trigonal Jahn-Teller splitting at the point is positive (namely the band originating from the state is higher in energy with respect to the twofold degenerate one arising from the and atomic orbitals) and very similar in magnitude for both systems, as it can be seen in Fig.2. Surprisingly, in selenides, despite a larger distortion, the apparent Jahn-Teller splitting is almost zero or negative.
This apparent contradiction can be solved by considering the hybridization between the transition metal t2g bands and the other occupied chalcogen bands at zone center (labeled “h” in Fig.2). In sulfides, this band is mainly formed by sulfur 3 states. In TaS2 this separation is larger than in NbS2, mainly due to the larger energy misalignment between the sulfur 3 and the Nb 4 or Ta 5 states. The situation is very different in selenides, where the hybridization between the Se states and the Ta or Nb states is strong (stronger in Nb than in Ta) and leads to completely counterintuitive results with respect to crystal field theory. For example, the larger octahedral distortion occurs in NbSe2, but here we find an apparent negative Jahn-Teller splitting. Finally, in TaSe2 the apparent Jahn-Teller splitting is almost zero as the crystal field and the hybridization perfectly cancels out and the t2g bands become almost threefold degenerate at zone center. Furthermore the top of what were the chalcogen -bands in sulfides becomes mixed with -states in selenides (particularly evident in NbSe2).
The different magnitude of the hybridization explains why in sulfides one expect t2g manifolds separated by the chalcogen states while in selenides the character is more entangled Calandra (2018).
Finally, it is worth mentioning that as the 1T-polytype breaks the inversion symmetry, we investigate the magnitude of relativistic effects in 1T-NbS2 finding them negligible, as expected given the relatively light atoms involved.
Having understood the electronic structure of the highly symmetric phase in comparison with other 1T compounds, we compare the energy of single layer 1T-NbS2 with the 1H-NbS2 polytype. We find that 1H is more stable by approximately mRy/Nb (see also Tab. 2), similarly to what happens for the NbSe2 caseCalandra and Mauri (2005); Kamil et al. (2018); Pasquier and Yazyev (2018). This large energy difference prevents an highly symmetric 1T phase to form in experiments.
In order to inspect for possible CDW instabilities, we then calculate the phonon dispersion of single layer 1T-NbS2. As shown in Fig.3 we found strongly unstable phonon modes. To better identify the wavevector of the most unstable phonon frequencies in the BZ, we also perform a 2D plot of the instability in Fig.3. At the harmonic level we find that the two most likely instabilities have wavevector compatible with a Ge and Liu (2010) and a CDWs.
III.2 Charge density wave phases
We perform geometrical optimization within the GGA approximation in and supercells starting from initial configurations obtained by displacing the atomic coordinates following the patterns of the most unstable phonon modes. In both case, we find structures that are substantially more stable than the highly symmetric ones. In the case of a supercell, we find two different reconstructions that are practically degenerate in energy (see Fig.4). Both CDW, however, seems to try to form some kind of star-of-David reconstruction, but the non ideal periodicity hinders the complete formation. This is confirmed by the fact that is the most stable reconstruction, with an energy gain of mRy/Nb with respect to the highly symmetric 1T-NbS2 phase, however still with an energy loss of mRy/Nb with respect to the highly symmetric 1H-NbS2 phase.
The optimized structure is shown in Fig.4 (we also report the Wyckoff positions in App. A), and it results dynamically stable (see App. B), the relative energy differences among the different phases considered are reported in Tab. 2.
The non-magnetic electronic structure in the phase is shown in Fig.5 (top). It is characterized by the presence of an extremely flat band at the Fermi level having a non-negligible character related to the central Nb atom in the star. The flat band is isolated from the others and is located in the middle of the gap, this is in analogy with what happens in 1T-TaS2Darancet et al. (2014); Miller et al. (2018); Albertini et al. (2016); Ge and Liu (2010); Liu (2009) and in contrast with the 1T-NbSe2 caseCalandra (2018); Pasquier and Yazyev (2018); Kamil et al. (2018) where the flat band is entangled with chalcogen states.
In order to disentangle the effects of chemistry and distortion in determining the energy position of the flat band with respect to the chalcogen states, we calculate the non-magnetic electronic dispersions of (i) NbS2 CDW structure in which we substitute the S atoms with Se keeping, however, the structure unchanged (labeled “1T-NbS2 str.; Se PP” in Fig.5), (ii) NbS2 using the crystal structure of 1T-NbSe2 in the CDW phase (labeled “1T-NbSe2 str.; S PP” in Fig.5). These calculations should be directly compared with the case of NbSe2 reported in Ref.Calandra (2018) (Fig.3, left panel) where, at the GGA level, the flat band lies in the middle of Se states and is not isolated from the others. This comes mostly from a 0.25 eV upshift of the lower occupied states at zone center.
Calculation (i) allows us to determine the effect of alignment between Se/S states with Nb ones. As it can be seen the effect of replacing the S with a Se pseudopotential is an up-shift of the Se states at zone center, in agreement with what happens in the ideal high-symmetry undistorted 1T-NbS2/1T-NbSe2 phases. However this up-shift is still not large enough to mix the flat band with the other occupied bands, as it happens in the phase of 1T-NbSe2Calandra (2018). If, on the contrary, we use the NbSe2 structure with the S pseudopotential, as in calculation (ii), we see that the results is to up-shift mostly the occupied states very close to the flat band. However, this is not what happens in the phase of 1T-NbSe2 as in this system, at the GGA level, the top of the Se states at are empty and are at higher energies then the flat band. It follows that the effect is not properly chemical neither structural, but it is a cooperative effect of the two. This aspect is a general feature strictly related to the chalcogen atom involved in the compound. In fact the same behavior is observable also in TaS2 and TaSe2 (see Refs.Calandra (2018); Darancet et al. (2014); Miller et al. (2018)).
As shown in Fig.5 (top), the Nb band is extremely flat, with a dispersion of eV. This implies a small Fermi velocity, a low kinetic energy and an high peak in the density of the states at the Fermi level. It is then natural to expect electronic instabilities to occur. We then perform spin-polarized calculations stabilizing an insulating ferrimagnetic solution with an energy loss of about 0.1 mRy/Nb with respect to the metallic non-magnetic solution (see Tab. 2). Thus, even in the absence of an Hubbard term, GGA stabilizes a magnetic state.
The magnetic bands are reported in Fig.6 (left), as we can see, in the ferrimagnetic configuration, the system results to be semiconductor with a gap of about 0.15 eV. The flat band is splitted in two, one is fully occupied and the second one is empty, so that the total magnetic moment is 1 . By referring to the left panel in Fig.4, the magnetic moments are 0.21 on the red sites, 0.04 on the blue ones and negligible contributions on the others. It is worth to underline that the magnetic structure is still unstable (4.4 mRy/Nb) with respect to the 1H one.
The insurgence of magnetism induces a weak hardening of some A2g modes, in principle detectable as a Raman shift (see App. B).
However, given the correlated nature of the problem and the key role of the DFT+U approximation in determining total energies, as shown in 1T-NbSe2Calandra (2018), we perform DFT+U calculations using the method in Ref. Cococcioni and de Gironcoli (2005). The parameter is computed self-consistently from first principlesCococcioni and de Gironcoli (2005), we obtain eV. This value is similar to those found for 1T-TaS2Darancet et al. (2014) and 1T-NbSe2Calandra (2018) compounds.
We first perform structural optimization of the high-symmetry 1T-NbS2 and 1H-NbS2 single-layer structure within DFT+U. For the 1H-polytype, we find that the in-plane lattice parameter in DFT+U is in slightly better agreement with the one measured in the bulk than in the GGA case (see Tab. 2), suggesting that DFT+U gives a slightly better energetic then GGA, as it happens in NbSe2Calandra (2018). As it can be seen, the energy difference between the 1H and 1T undistorted polytypes is now reduced.
We then optimize the geometry in the CDW phase with DFT+U obtaining a small contraction of the in-plane lattice constants (see Tab. 2, the Wyckoff positions are reported in App. A). Also the magnetic structure is slightly different from GGA results: a stronger ferrimagnetic solution with magnetic moments of 0.41 on the red sites, 0.03 on the blue ones and negligible antiferromagnetic contributions from the other sites is stabilized (we refer to the left panel in Fig.4 for color labeling). The total magnetic moment per unit cell is of 1 . The magnetic moment on the central atom is thus almost the double of the one found in spin-polarized GGA (while the total spin is of course still ). Moreover, as shown in Tab. 2, the CDW magnetic solution has an important energy gain with respect to the 1T-polytype and it is almost degenerate with the 1H one (0.5 mRy energy difference).
This energy difference is slightly smaller than the one found between 1H-NbSe2 highly symmetric polytype and the 1T-NbSe2 charge density wave phase. This suggest that 1T-NbS2 can be synthesized with a similar experimental procedure to the one used for NbSe2 Nakata et al. (2016); Bischoff et al. (2017).
In Fig.6 we report the band structure for the magnetic solution obtained in DFT+U compared with the ones obtained in GGA. The band-gap between the flat band is now more than twice that in spin-polarized GGA (0.41 eV) and is not the fundamental gap as the minority spin flat bands is pushed inside the empty -conduction bands. The fundamental gap is 0.35 eV and involves transitions between -orbitals of different Nb atoms.
Finally we evaluate the nearest-neighbor () and next-near-neighbor () exchange constants between different star of David clusters in an ferromagnetic Hubbard model described by the following Hamiltonian:
[TABLE]
We adopted a super-cell approach and considered a cell with different collinear magnetic configurations. It is important to note that we obtain similar total energies for all the spin configurations took into account.
The calculated ferromagnetic exchange couplings are =9.5 K and =0.4 K, in line with the parameters describing the similar NbSe2 compoundPasquier and Yazyev (2018). From that the system results to have a ferromagnetic ground state between different stars.
IV Conclusions
In this work we investigated by first principles the possible formation of single-layer 1T-NbS2 as well as its structural, electronic and dynamical properties in the high symmetry phase and in the CDW one with different degrees of correlation and allowing for magnetic solutions.
We demonstrate that the 1T undistorted () polytype is highly unstable towards a reconstruction. Within the GGA+U approximation, the structural distortion and the formation of a ferrimagnetic state cooperate in stabilizing the 1T-NbS2 phase in single layer form that becomes comparable in energy with that of the 1H polytype. Thus, we predict that this system can be synthesized with similar techniques to those used for single layer 1T-NbSe2Nakata et al. (2016); Bischoff et al. (2017). Interestingly, a previous workCarmalt et al. (2004) describes growth of bulk 1T-NbS2 on glass keeping the substrate at very high temperature. As a similar technique has been used for the synthesis of 1T-NbSe2 (with a different substrate) in Ref. Nakata et al. (2016), this makes the synthesis of single layer 1T-NbS2 even more likely.
Finally, it is interesting to underline that in this system, magnetism occurs in a ultraflat band, isolated from all the others and having a marked d character on the central Nb atom in the star. Spin polarized calculations without any Hubbard mean field term, recover the insulating state by stabilizing magnetism, (although with a fairly small gap). A similar effect occurs in TaS2Darancet et al. (2014), where even at a magnetic state is stabilized within the spin polarized generalized gradient approximation. In this respect, sulfides are odd with 1T-NbSe2 where the flat band is strongly hybridized with Se states and the Hubbard interaction is needed to disentangle it from the other bandsCalandra (2018). 1T-NbS2 in the is then a prototype system where the presence of an ultraflat band produces magnetism even at very moderate values of .
Acknowledgments
We thank G. Menichetti for useful and stimulating discussions.
This work was supported by French state funds managed by the ANR within the Investissements d’Avenir program under references ANR-13-IS10-0003-01 ANR-11-IDEX-0004-02, and more specifically within the framework of the Cluster of Excellence MATISSE led by Sorbonne Universitè, by the European Graphene Flagship (GrapheneCore 2). Computer facilities were provided by CINES, IDRIS, and CEA TGCC and PRACE (2017174186).
Appendix A
We report the relaxed Wyckoff positions for the low temperature CDW (non-magnetic) phase. Both structures obtained in GGA and GGA+U belongs to the space group (group number 147), and the Wyckoff positions are reported in Tab. 3 and Tab. 4 respectively.
Appendix B
We compute the phonon dispersion for the CDW phase in GGA approximation for the non magnetic (NM) and the ferrimagnetic (FM) solutions. We calculate the dynamical matrix at zone center and then Fourier interpolate the dynamical matrices on the full Brillouin zone. Results are reported in Fig.7. All frequencies are positive revealing the CDW phase is dynamically stable. The Raman active modes are reported in Tab. 5. The insurgence of magnetism causes a weak hardening of A2g frequencies at around 90 and 366 cm*-1* (we register shifts of about cm*-1*, see Tab. 5).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Wilson et al. (1975) J. A. Wilson, F. J. Di Salvo, and S. Mahajan, Adv. in Phys. 24 , 117 (1975).
- 2FAZ (1980) Physica B+C 99 , 183 (1980) . · doi ↗
- 3Fazekas and Tosatti (1979) P. Fazekas and E. Tosatti, Phyl. Mag. B 39 , 229 (1979).
- 4Darancet et al. (2014) P. Darancet, A. J. Millis, and C. A. Marianetti, Phys. Rev. B 90 , 045134 (2014) . · doi ↗
- 5Ngankeu et al. (2017) A. S. Ngankeu, S. K. Mahatha, K. Guilloy, M. Bianchi, C. E. Sanders, K. Hanff, K. Rossnagel, J. A. Miwa, C. B. Nielsen, M. Bremholm, and P. Hofmann, Phys. Rev. B 96 , 195147 (2017).
- 6Carmalt et al. (2004) C. J. Carmalt, T. D. Manning, I. P. Parkin, E. S. Peters, and A. L. Hector, J. Mater. Chem. 14 , 290 (2004).
- 7Novoselov et al. (2005) K. S. Novoselov, D. Jiang, F. Schedin, T. Booth, V. V. Khotkevich, S. Morozov, and A. K. Geim, PNAS 102 , 10451 (2005).
- 8et al. (2011) J. N. C. et al. , Science 331 , 568 (2011).