Experimental and molecular dynamics study of the ionic conductivity in aqueous LiCl electrolytes
Are Yll\"o, Chao Zhang

TL;DR
This study combines experimental measurements and molecular dynamics simulations to analyze ionic conductivity in aqueous LiCl electrolytes, revealing that at high concentrations, Li+ and Cl- transference numbers become comparable, independent of force fields.
Contribution
It provides a combined experimental and simulation analysis of ion conduction in LiCl solutions, highlighting concentration-dependent mobility effects.
Findings
Transference numbers of Li+ and Cl- become comparable at high concentration.
Results are consistent across different force fields.
Ion mobility depends on concentration, affecting conductivity.
Abstract
Lithium chloride LiCl is widely used as a prototype system to study the strongly dissociated 1-1 electrolyte solution. Here, we combined experimental measurements and classical molecular dynamics simulations to study the ion conduction in this system. Ionic conductivities were reported at both 20C and 50C from experiments and compared to results from molecular dynamics simulations. The main finding of this work is that transference numbers of Li and Cl become comparable at high concentration. This phenomenon is independent of the force fields employed in the simulation and may be resulted from the ion-specific concentration dependence of mobility.
Click any figure to enlarge with its caption.
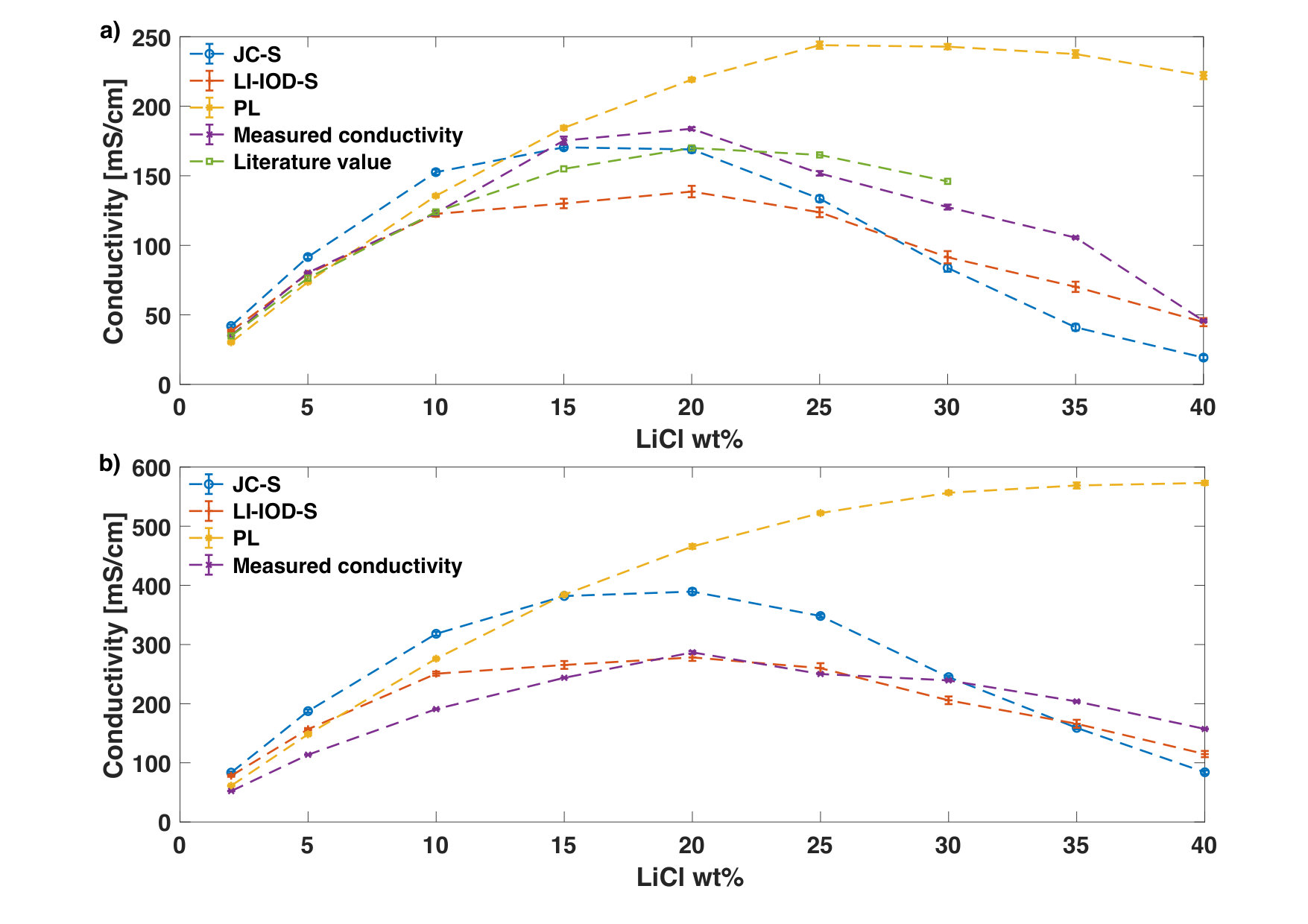 Figure 1
Figure 1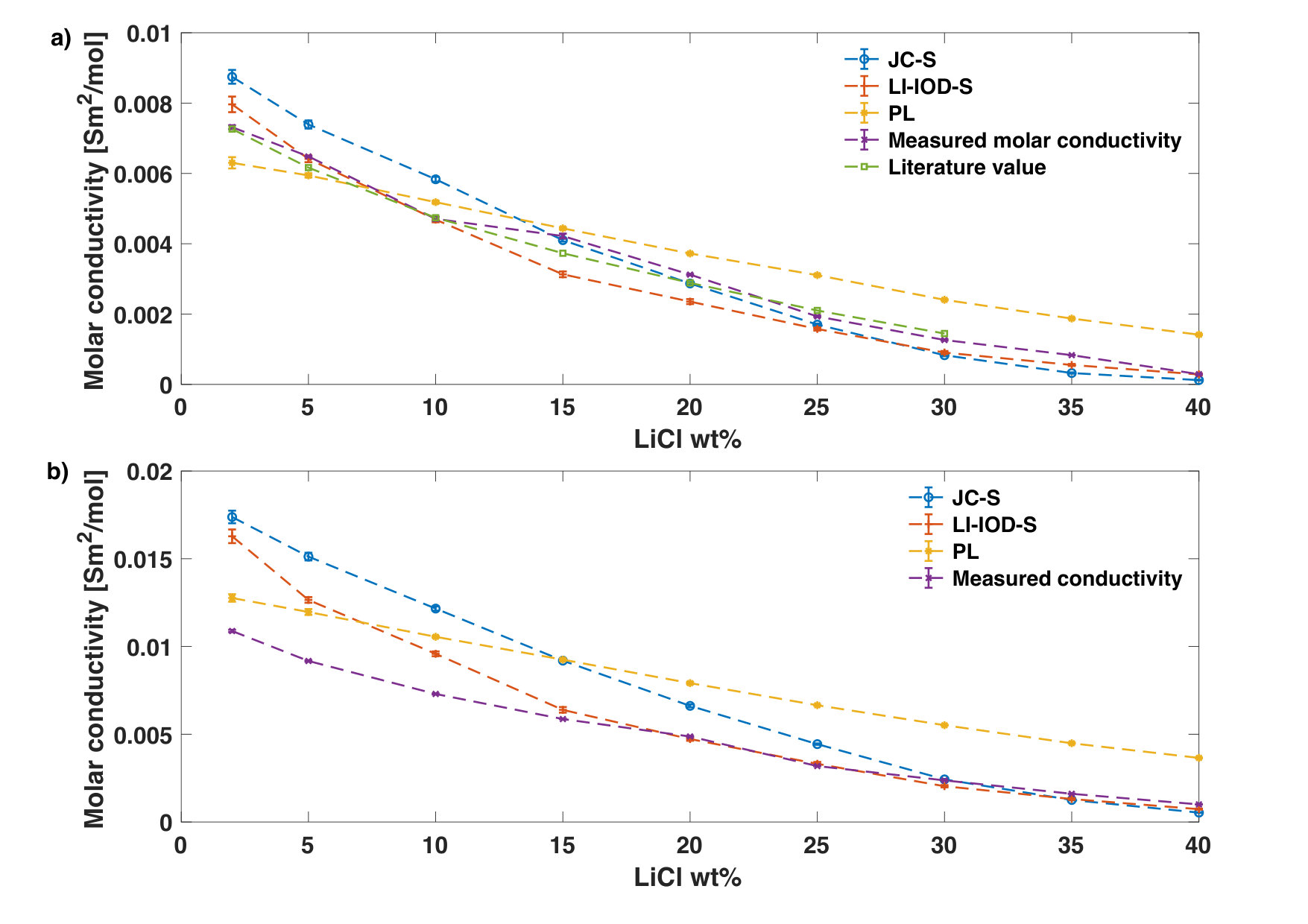 Figure 2
Figure 2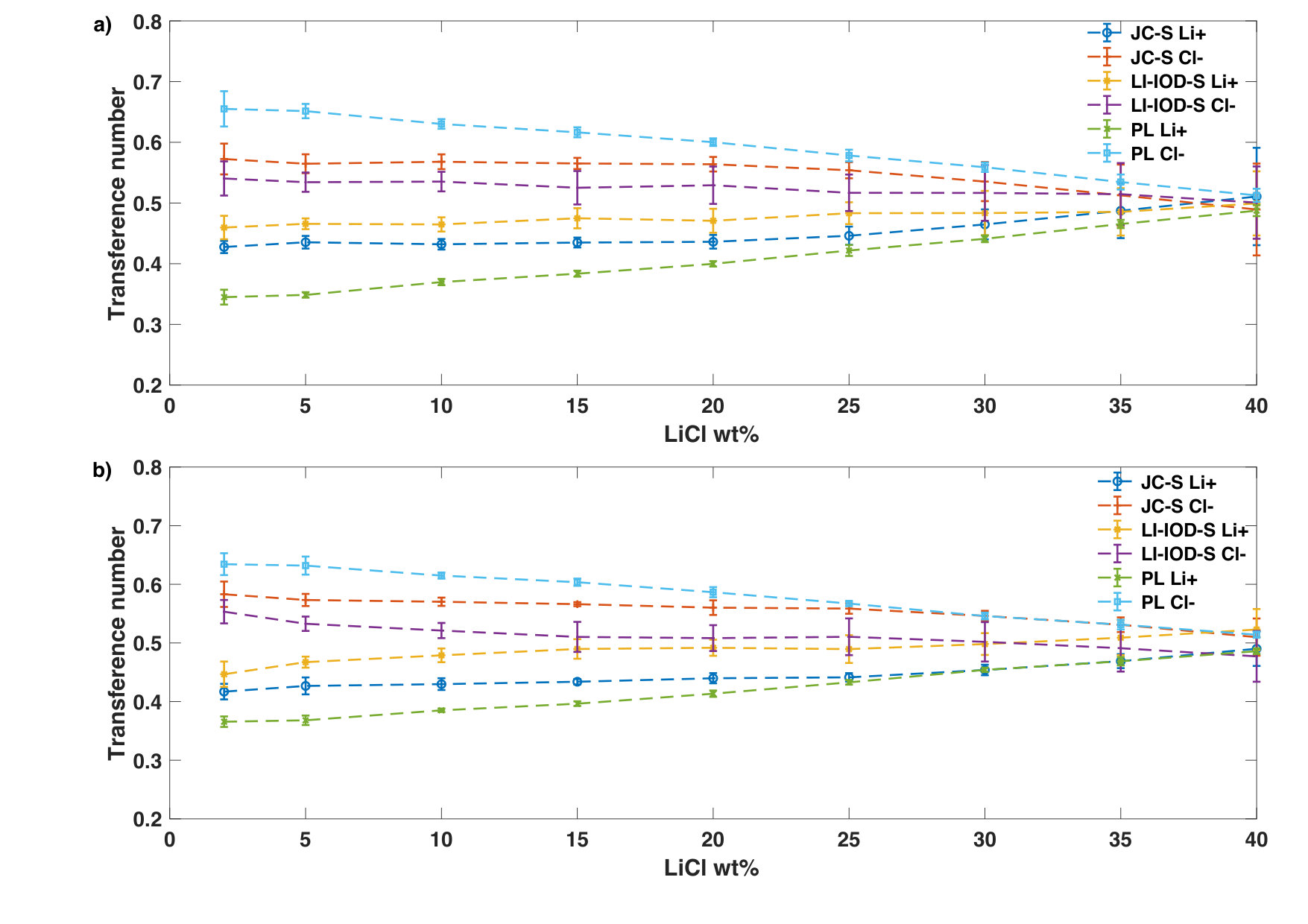 Figure 3
Figure 3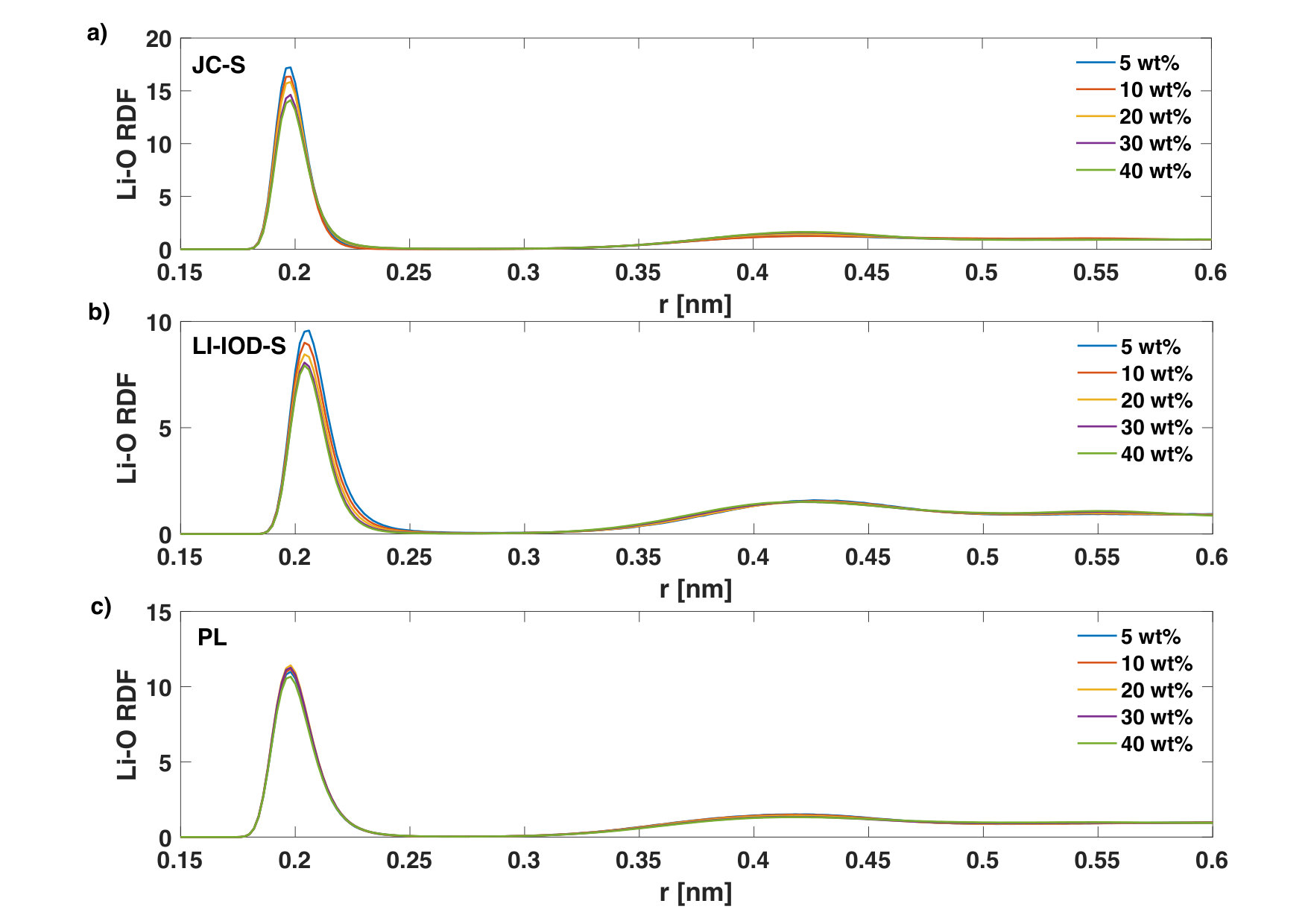 Figure 4
Figure 4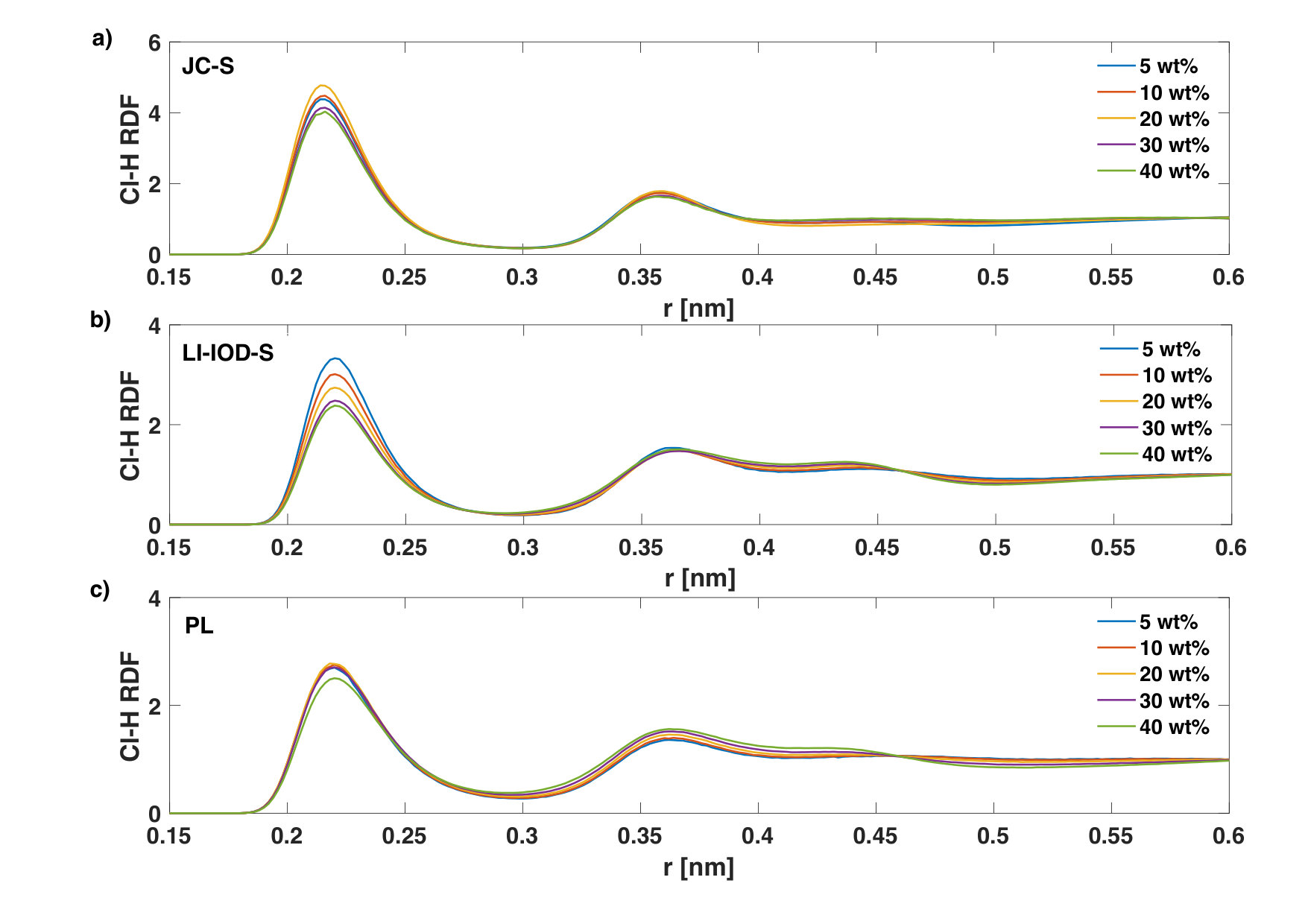 Figure 5
Figure 5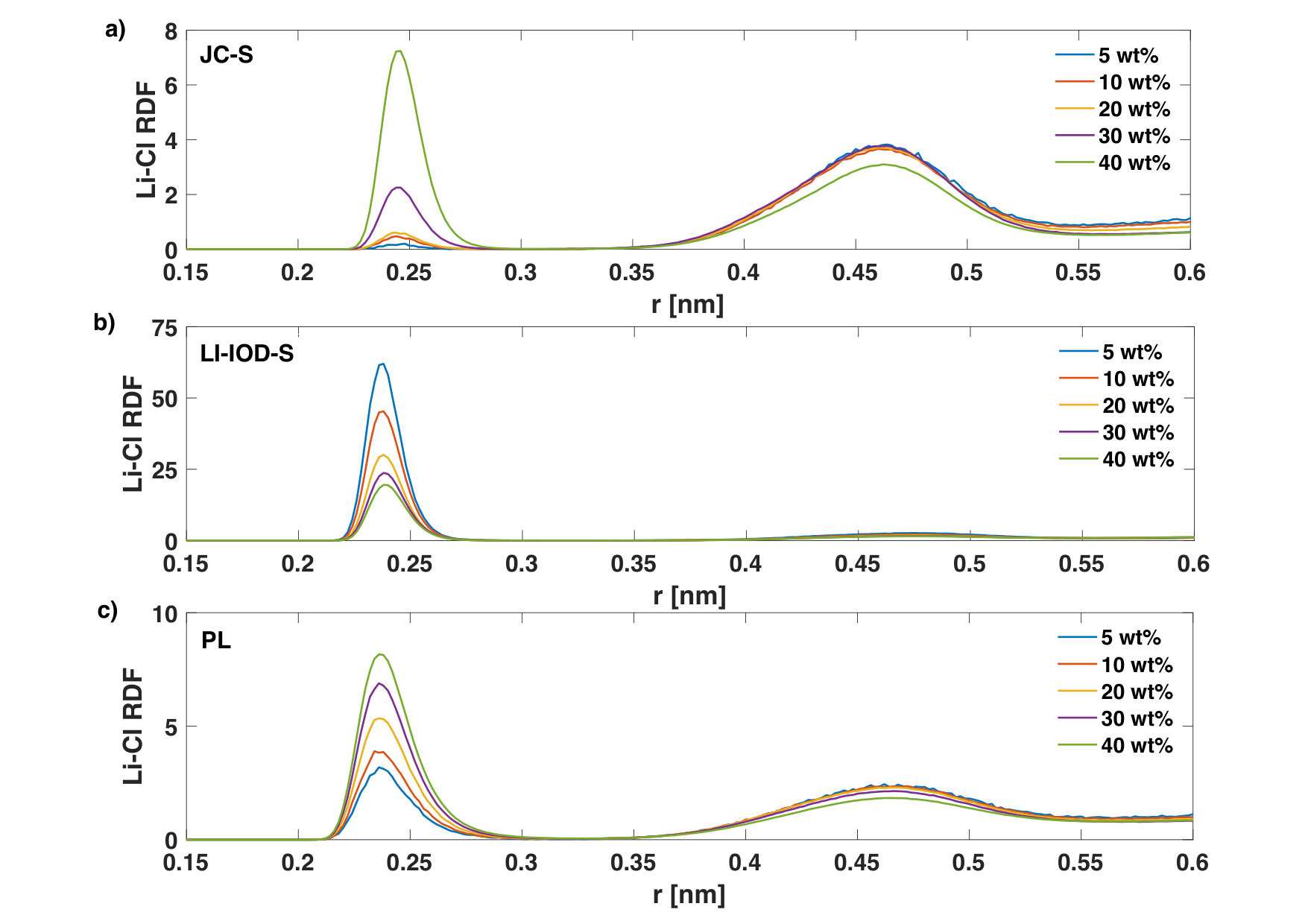 Figure 6
Figure 6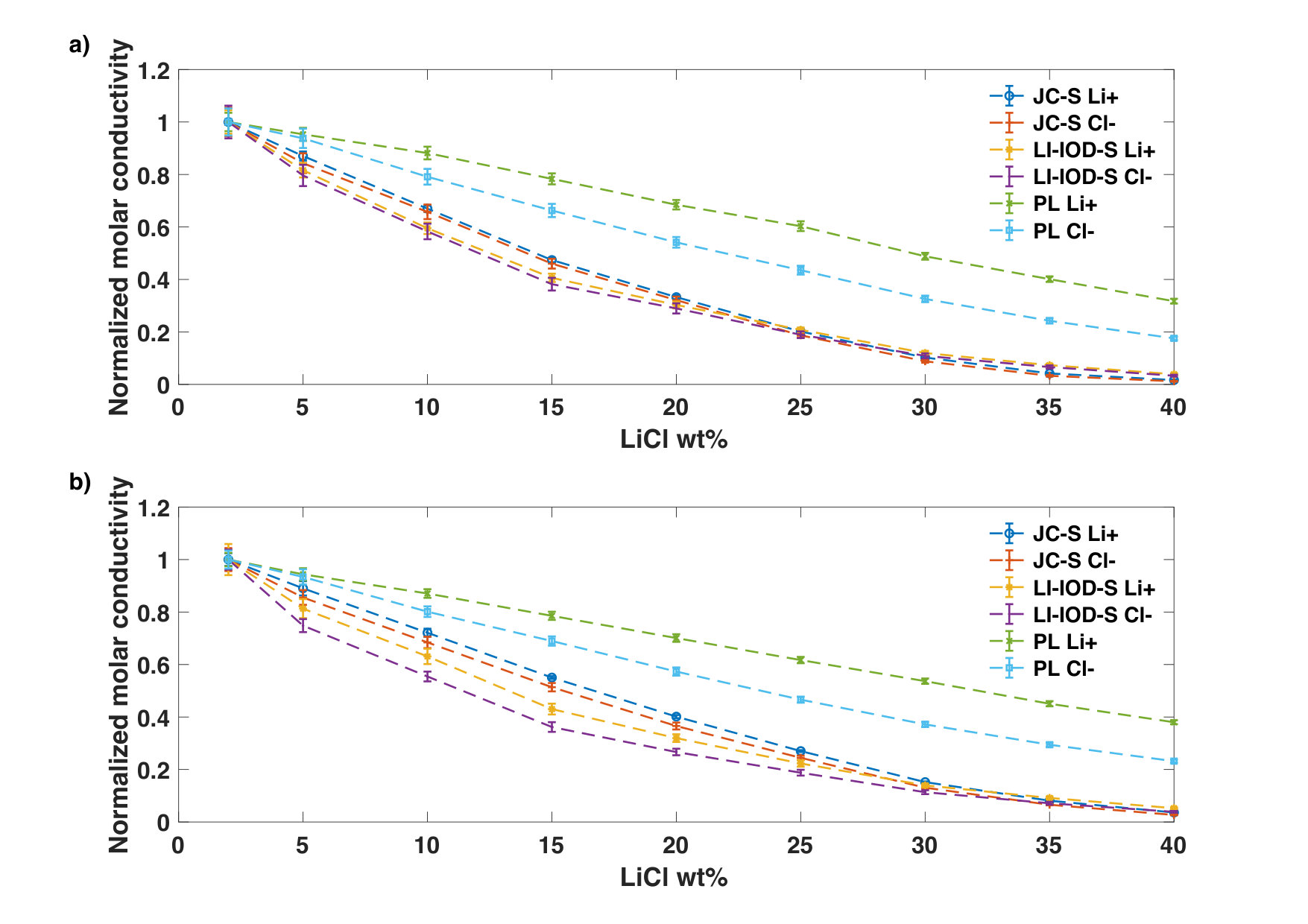 Figure 7
Figure 7| wt% LiCl | 5 | 10 | 20 | 30 | 40 |
|---|---|---|---|---|---|
| JC-S: Li+-O | 4.19 | 4.16 | 4.10 | 3.87 | 3.2 |
| 4.20 | 4.17 | 4.08 | 3.83 | 3.2 | |
| LI-IOD-S: Li+-O | 3.73 | 3.30 | 2.79 | 2.34 | 2.0 |
| 3.63 | 3.2 | 2.67 | 2.27 | 2.0 | |
| PL: Li+-O | 3.95 | 3.88 | 3.65 | 3.28 | 2.8 |
| 3.92 | 3.82 | 3.58 | 3.19 | 2.7 | |
| JC-S: Cl--H | 6.81 | 6.84 | 6.85 | 6.57 | 5.4 |
| 6.65 | 6.67 | 6.64 | 6.37 | 5.3 | |
| LI-IOD-S: Cl--H | 5.88 | 5.29 | 4.50 | 3.79 | 3.3 |
| 5.65 | 5.06 | 4.18 | 3.63 | 3.1 | |
| PL: Cl--H | 5.56 | 5.50 | 5.11 | 4.65 | 3.9 |
| 5.29 | 5.27 | 4.90 | 4.32 | 3.6 | |
| JC-S: Li+-Cl- | 0.00 | 0.01 | 0.03 | 0.18 | 0.8 |
| 0.01 | 0.02 | 0.07 | 0.24 | 0.8 | |
| LI-IOD-S: Li+-Cl- | 0.62 | 0.96 | 1.35 | 1.75 | 2.0 |
| 0.67 | 1.01 | 1.46 | 1.81 | 2.1 | |
| PL: Li+-Cl- | 0.05 | 0.12 | 0.35 | 0.71 | 1.2 |
| 0.07 | 0.16 | 0.4 | 0.78 | 1.3 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Experimental and molecular dynamics study of the
ionic conductivity in aqueous LiCl electrolytes
Are Yllö
Chao Zhang
Department of Chemistry-Ångström Laboratory, Uppsala, University, Lägerhyddsvägen 1, 75121 Uppsala, Sweden
Abstract
Lithium chloride LiCl is widely used as a prototype system to study the strongly dissociated 1-1 electrolyte solution. Here, we combined experimental measurements and classical molecular dynamics simulations to study the ion conduction in this system. Ionic conductivities were reported at both 20∘C and 50∘C from experiments and compared to results from molecular dynamics simulations. The main finding of this work is that transference numbers of Li+ and Cl- become comparable at high concentration. This phenomenon is independent of the force fields employed in the simulation and may be resulted from the ion-specific concentration dependence of mobility.
keywords:
Electrolyte solution , Ionic conductivity , Molecular dynamics , Force fields , Debye-Onsager theory
1 Introduction
Aqueous electrolytes play important roles in many areas of science and engineering, such as electrophysiology, electrochemistry and colloid science. Simple 1-1 electrolyte which is completely dissociated in dilute solution is often used as a prototype system to develop analytical theories such the well-known Debye-Hückel theory [1]. This tradition dates back to the beginning of Physical Chemistry and coins the early physical chemists as “Ionists” [2].
Lithium chloride (LiCl) as an example of these simple 1-1 electrolytes is of particular interest due to its very high solubility ( 45 wt% at room temperature). The structure of LiCl solution has been extensively investigated by X-ray diffraction and neutron scattering experiments [3, 4, 5] in together with reverse Monte Carlo and molecular dynamics simulations [6, 7, 8, 6, 9, 10, 11, 12]. The synergy between experiments and simulations has been proven to be useful to gain a deeper understanding of solvation structures of Li+ and Cl-.
In the molecular dynamics simulation community, another interest of modeling LiCl solution was on developing various kinds of force-fields where cations and anions are commonly described by Lennard-Jones (LJ) potential and point charge [13, 14]. Despite of its simplicity, this approach has been shown be capable to capture both single ion properties (such as the hydration free energy) to ion-ion interactions as reflected in radial distribution functions and the solubility [11]. We refer interested readers to a recent work on this topic for a comprehensive overview and benchmarks [12].
On the other hand, the dynamical and transport properties of these models were often overlooked. In particular, the ionic conductivity of LiCl calculated from molecular dynamics simulations has not been compared to experimental measurements at both room temperature and elevated temperature. This fact is somehow surprising, because the basic function of any electrolyte is to serve as an ionic conductor.
In this work, we carried out both experimental measurements and molecular dynamics simulations of the ionic conductivity in LiCl solutions. Ionic conductivities were reported at both 20∘C and 50∘C from experiments and compared to those calculated from molecular dynamics simulations using three different force-field models [10, 13, 14] (See Section 2 for details) and SPC/E water [15]. In addition to provide reference data for future force-field developing works, the main finding of our study is that transference numbers (i.e. the fractional contribution to the ionic conductivity) of Li+ and Cl- become comparable at high concentration. This phenomenon is independent of the force fields employed in the simulation and can be explained by taking into account the ion-paring and ion-specific effects. The later imposes a challenge to the Debye-Onsager theory of the ionic conductivity.
2 Experimental and computational methods
2.1 Ionic conductivity measurements
The conductivity measurement of LiCl at 2, 5, 10, 15, 20, 25, 30, 35 and 40 wt % were performed with an ”InLab” conductivity meter (Mettler Toledo). The conductivity meter probe used is a 4 pole InLab 738-ISM by (Mettler Toledo) which has a sensitivity range from 0.01–1000 mS/cm and gives accurate measurements up to 100∘C. Before measuring, the probe was calibrated with a standardized 12.88 mS/cm potassium chloride (KCl) solution (Mettler Toledo). After the successful calibration of the instrument, the probe was lowered into respective solution. The measurement ran until both the conductivity and the temperature of the solution had equilibrated at a stable value. The mean of the five independent measurements were then noted as the final conductivity of that solution at 20∘C.
Similar measurements were then done at an elevated temperature of approximately 50∘C. The solutions were heated to 50∘C by placing them in a heated water bath with an external thermometer attached to a reference plastic container with deionized water. When the solution had reached the sought-after temperature, the measurements were carried out in the same way as before.
2.2 Molecular dynamics simulations
The initial cubic box containing simple point charge/extended (SPC/E) water molecules [15] and random distributed Li+/Cl- ions was 2.963 nm for each side. Water molecules were kept rigid using the SETTLE algorithm [16]. The Ewald summation was implemented using the Particle Mesh Ewald (PME) [17] scheme and short-range cutoffs for the van der Waals and Coulomb interaction in the direct space are 1 nm.
Three force fields (ion models) for LiCl were chosen in this study which are Joung-Cheatham III (JC-S) [13], Li-Song-Merz (LI-IOD-S) [14] and Pluhařová -Mason-Jungwirth (PL) [10]. JC-S was parameterized against thermodynamic data such hydration free energy and lattice energy of salt crystal and has been validated for higher salt concentration [18, 19] at room temperature. LI-IOD-S focus on the structural aspect and was fitted to the ion oxygen distance in the first solvation shell. PL-S was tuned by scaling down the point charge of each ion by the refractive index of liquid water in order to make up the missing electronic polarization. The corresponding LJ parameters and point charges of these three models are summarized in Table 1. In all cases, the Lorentz-Berthelot combination rule was used between two dissimilar non-bonded atoms.
Regarding the technical setting in simulations, the steepest descent algorithm was used for the energy minimization before the equilibration. The NVT (constant number of particles, constant volume and constant temperature) equilibration ran for 1 ns with the timestep of 2 fs. The temperature was then held in place using the Bussi-Donadio-Parrinello thermostat which preserves both thermodynamic and dynamic properties [20]. The follow-up NPT (constant number of particles, constant pressure and constant temperature) simulations ran for 10 ns each and trajectories were collected every 0.5ps for conductivity calculation and structural analysis. During the NPT simulations, Parrinello-Rahman barostat [21] was employed with a reference pressure of 1.0 bar. This simulation protocol was used for LiCl solution at 2, 5, 10, 15, 20, 25, 30, 35 and 40 wt % and both 20∘C and 50∘C and all simulations were performed using GROMACS 4 package [22]. This corresponds to simulations with following compositions in terms of number ratio /: 6/694, 15/676, 30/646, 46/614, 61/584, 77/552, 94/518, 110/486, 127/452. Statistical errors were estimated using the standard deviation of observables from 5 equispaced segments in the full trajectory.
3 Results and discussion
3.1 Ionic conductivity and transference number
The simplest way to calculate the ionic conductivity in molecular dynamics simulations is to use the Nernst-Einstein equation [23]:
[TABLE]
where is the ionic conductivity of the solution, and are ionic conductivities for cation and anion respectively. and are point charges of ions in the model. is the number density of the salt, is Boltzmann constant and is the temperature.
and are self-diffusion coefficients of cation and anion respectively and computed from the corresponding mean squared displacement using the Einstein relation as follows:
[TABLE]
where is the time, is the number of LiCl salt, is the position of th cation or anion, indicates the ensemble average.
One should note that the the Nernst-Einstein equation holds only for non-interacting charged particles in a homogeneous and isotropic solvent. Thus, the ion-ion correlation is not taken into account in the formula. In other words, the ionic conductivity calculated using the Nernst-Einstein equation gives an upper bound of the actual value.
On the other hand, since is a sum of individual contributions of cations and anions by construction, the transference number can be readily extracted as:
[TABLE]
From Fig. 1a, we see that the results from JC-S is the one that comes closest to the measured and the literature values in the whole concentration range at 20∘C, although three ion models seem be equally well at lower concentrations. Our measured molar conductivity is also in accord with the result in a recent report [25]. At 50∘C (See Fig. 1b), LI-IOD-S gives results which agrees best with measured values. JC-S overestimates the conductivity for lower to mid-range concentrations and underestimates it for higher concentrations. At both temperatures, PL significantly overestimates the conductivity from mid to high concentrations. Similar behavior of PL has been reported for the diffusion coefficient of Li+ and Cl- recently [12]. This is likely due to the fact that point charge of ions are scaled down in this model which leads to a much weaker ion-solvent interaction.
Both JC-S and LI-IOD-S manage to describe the parabola behavior of the ionic conductivity as a function of the concentration and to provide accurate estimates of the corresponding concentration at the conductivity maximum. The reason for the conductivity maximum comes from a tradeoff between the increase of number of charge transportors and the decrease of their mobility as the concentration goes up. When molar conductivities are plot instead (Fig. 2), one can see clearly that the mobility of ions reduces as a function of the concentration. Results of JC-S and LI-IOD-S have better agreements with experiments while PL shows a much higher deviation in the mid-to-high concentration range.
Fig. 3 shows the transference numbers of Li+ and Cl- of three models at both 20∘C and 50∘C. The chloride ions contributes a larger fraction of the electrical current (0.55 to 0.65) while lithium ions stand for a smaller fraction (0.45 to 0.35). However, this gap diminishes as the concentration increase and eventually the transference numbers become similar nearly the solubility limit.
3.2 Radial distribution function and ion-pairing
The configurational distribution function can be reduced to its two-particle version as [26]:
[TABLE]
which gives the joint probability distribution to find one particle at position and any other particle at . Note that the factor accounts for all possible pairs.
In an ideal gas, particles are uncorrelated. As a result, the simply equals to where is the number density. This leads to the definition of the quantity called the pair distribution function:
[TABLE]
This quantity reflects the density deviation from the (uncorrelated) ideal gas.
For isotropic fluid, this function depends upon , this makes called a radial distribution function.
The coordination number, i.e. the number of neighbouring atoms within first minimum of the from a central atom, is define as:
[TABLE]
The first peak of steadily decreases for both JC-S and LI-IOD-S ion models at 20∘C with increasing LiCl concentration (Fig. 4). Similar trend was seen for with LI-IOD-S (Fig. 5). This is expected, since the coordinating hydrogen/oxygen atoms of water molecules are gradually replaced by the counter-ions, see Table 2. The anomaly is that at 20wt% has the highest first peak with JC-S. In the case of PL, the peak heights of and are not much modulated by the concentration.
Regarding the radial distribution function of Li+-Cl-, it goes up with increasing concentration (Fig. 6) for JC-S and PL. An opposite trend was found in the case of LI-IOD-S. Despite that, coordination numbers between Li+ and Cl- become larger with the concentration for all three ion models, which is a sign of ion-pairing.
We notice that the coordination number of Li+-O and Cl--H in LI-IOD-S is more sensitive to the concentration, in contrast to other two ion models (Table 2). This is in accord with Fig. 4 and Fig. 5.
Although radial distribution functions at 50∘C have a similar concentration dependence (data not shown), coordination numbers of ion-water become smaller in most cases as shown in Table 2 which were expected, because hydration shells become less structured at elevated temperature. In contrast, all three ion models show that the cation-anion coordination number goes up with the temperature. This may be due to the fact that the dielectric constant of liquid water decreases with the temperature and the solvent screening is weaker accordingly.
3.3 Ion-paring contribution to the ionic conductivity
We mentioned at the beginning of Section 3.1 that the ionic conductivity calculated from the Nernst-Einstein equation provides an upper bound and the actual conductivity is always smaller because of the ion-paring (ion-ion correlation). Near the solubility limit, the ionic conductivity may be reduced by 30% when ion-ion correlations are taken into considered in the calculation [27]. This is similar to our estimation based on the mean square charge displacement [28], which gives a value of 40% for LiCl. Therefore, the overshooting of PL in the ionic conductivity at high concentration as shown in Fig. 1 is not because of the missing of ion-pairing contribution in Eq. 2 but likely due to the down-scaling of the charge in the model (See Table 1).
One of the main observations in this study is that the transference number of the chloride ion becomes similar to that of the lithium ion. This is in accord to the tracer diffusion measurement reported in the literature [29]. The standard explanation for this phenomenon is that lithium and chloride ions pair up at high concentration and move together in a concerted manner.
Instead, we notice that the relative reduction of the Cl- conductivity with the increase of the concentration can be notably larger than that of the Li+ conductivity in the case of PL (Fig. 7). . This observation is interesting because the PL model was adjusted to take into account the missing electronic polarization. In addition, we noticed that the difference in the concentration dependence between Li+ and Cl- becomes more apparent at 50oC because the dielectric constant goes down. Thus, it would be of interest to investigate the effect of polarization on the ion-specific concentration dependence of mobility.
The implication of this observation is twofold. Firstly, since the molar conductivity of the chloride ion at infinite dilution is larger than that of the lithium ion, therefore the ion-specific concentration dependence could lead to a crossover between cation transference numbers and anion transference number even without considering the ion-pairing. Secondly, the standard Debye-Onsager theory may need be expanded in order to consider ion-specific concentration dependence for mobility of Li+ and Cl-.
4 Conclusion
In this work, we carried out both experimental measurements and molecular dynamics simulations of the ionic conductivity in LiCl solutions. Ionic conductivities were reported at both 20∘C and 50∘C from experiments and compared to those calculated from molecular dynamics simulations using three different ion models. In addition to provide reference data for future force-field developments, the main finding of our study is that transference numbers of Li+ and Cl- become similar at high concentration. This phenomenon is independent of the force fields employed in the simulation and may be due to the ion-specific concentration dependence of mobility.
5 Acknowledgments
C.Z. thanks Uppsala University for a start-up grant. Funding from the Swedish National Strategic e-Science program eSSENCE is also gratefully acknowledged.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1[1] W. R. Fawcett, Liquids, solutions, and interfaces from classical macroscopic descriptions to modern microscopic details, Oxford University Press, Oxford; New York, 2004.
- 2[2] J. W. Servos, Physical chemistry from Ostwald to Pauling : the making of a science in America, Princeton Univ. Press, Princeton, NJ, 1996.
- 3[3] R. H. Tromp, G. W. Neilson, A. K. Soper, Water structure in concentrated lithium chloride solutions, J. Chem. Phys. 96 (11) (1992) 8460–8469.
- 4[4] K. Winkel, M. Seidl, T. Loerting, L. E. Bove, S. Imberti, V. Molinero, F. Bruni, R. Mancinelli, M. A. Ricci, Structural study of low concentration Li Cl aqueous solutions in the liquid, supercooled, and hyperquenched glassy states, J. Chem. Phys. 134 (2) (2011) 024515–9.
- 5[5] S. Ansell, A. C. Barnes, P. E. Mason, G. W. Neilson, S. Ramos, X-ray and neutron scattering studies of the hydration structure of alkali ions in concentrated aqueous solutions, Biophys. Chem. 124 (3) (2006) 171–179.
- 6[6] K. Ibuki, P. A. Bopp, Molecular dynamics simulations of aqueous Li Cl solutions at room temperature through the entire concentration range, J. Mol. Liq. 147 (1-2) (2009) 56–63.
- 7[7] I. Harsányi, L. Pusztai, On the structure of aqueous Li Cl solutions, J. Chem. Phys. 122 (12) (2005) 124512–7.
- 8[8] L. Petit, R. Vuilleumier, P. Maldivi, C. Adamo, Ab Initio Molecular Dynamics Study of a Highly Concentrated Li Cl Aqueous Solution, J. Chem. Theory. Comput. 4 (7) (2008) 1040–1048.