Supplementary Information for:
Design of a multifunctional polar metal via
first-principles high-throughput structure screening
Yue-Wen Fang1,2
[email protected]
Hanghui Chen2,3
[email protected]
1Department of Materials Science and Engineering, Kyoto University, Kyoto, Japan
2NYU-ECNU Institute of Physics, New York University Shanghai China
3Department of Physics, New York University, New York 10003, USA
Supplementary Note 1 Low-energy structures predicted from CALYPSO search
Supplementary Table 1. A list of predicted crystal structures including
ten lowest energy states, perovskite anti-polar state with Pmma symmetry,
and post-perovskite anti-polar state with Pmmm symmetry.
‘—’ represents non-perovskite and non-post-perovskite structures.
Post-perovskite structures are explicitly
shown. ‘Layered’, ’Rock-salt’, and ‘Columnar’ refer to different cation
orderings of A-site ordered double perovskite structure (the naming convention
follows Ref. B926757C ).
In our structure search, we consider all possible cation orderings in
perovskite structure, as well as non-perovskite structures such as
post-perovskite and hexagonal structures.
Supplementary Table LABEL:tab:structureinfo lists twelve crystal structures of
BiPbTi2O6, including ten lowest energy structures predicted by
CALYPSO, perovskite anti-polar structure with Pmma symmetry and
post-perovskite anti-polar structure with Pmmm symmetry. All the
energies in Supplementary Table LABEL:tab:structureinfo are normalized to per
formula unit (i.e., 10-atom BiPbTi2O6). The total energy of
post-perovskite Pmm2 structure (the one with the lowest energy) is
set as the zero point.
Supplementary Note 2 Structural information of available substrates
Supplementary Table 2. Structural information of perovskite oxide substrates KTaO3 and
NdScO3, calculated by DFT-PBEsol method. The experimental lattice constants,
taken from Ref. BISWAS2017117 are also shown in the parentheses
for comparison.
We study two perovskite oxide substrates KTaO3 and
NdScO3 BISWAS2017117 . As shown in
Supplementary Table LABEL:tab:substrate, the DFT (PBEsol) calculated lattice
constants of KTaO3 and NdScO3 are in good agreement with the
experimental lattice constants (within 1% difference). We find
that cubic perovskite KTaO3 (cubic lattice constant of ∼ 4.00
Å) and orthorhombic perovskite NdScO3 (pseudo-cubic lattice
constant of ∼ 4.08 Å) can impose tensile strain sufficiently to
stabilize perovskite Pmm2 BiPbTi2O6 in thin film form. In our
main text, the heterostructure of BiPbTi2O6/PbTiO3 is simulated to growth
on the substrate of KTaO3.
Supplementary Note 3 Pressure and strain study including P1 and I4mm structures
In this section, we study more crystal structures under pressure and strain.
We consider not only the three lowest energy structures (post-perovskite
Pmm2, perovskite Pmn21 and perovskite Pmm2), but also the other
two low-energy structures (a non-perovskite P1 and perovskite
I4mm). Panel a of Supplementary Figure 1 shows
the pressure dependence and panel b of Supplementary Figure 1
shows the strain dependence. The conclusion in the main text
does not change after we consider more low-energy structures.
Supplementary Note 4 Temperature effect on the phase transitions
In order to investigate the temperature effect on the phase transitions between
post-perovskite Pmm2, perovskite Pmn21 and perovskite Pmm2,
we study the Helmholtz free energies of the three phases.
The Helmholtz free energy for a defect-free non-magnetic system with atomic volume V at temperature T
can be approximated as Bansal2016 ; 1999Grimvall ; ZHU201411
[TABLE]
where Fph(V,T) is the phonon free energy (i.e., vibrational free energy) and Fele(V,T) is the thermal
electron contribution to the free energy.
The electron free energy Fele(V,T)
can be divided into the total energy E0 at 0 K and the remaining part Fˉele(V,T) PhysRevB.79.134106 :
[TABLE]
E0 can be calculated by standard density functional theory (DFT). In
quasi-harmonic approximation, Fˉele(V,T) can be
calculated by using Mermin’s finite temperature formulation of
DFT PhysRev.137.A1441 ; PhysRevB.79.134106 , but requires
very large supercell calculations (for BPTO, the supercells need to
contain 160∼320 atoms) with a range of volumes under the
studied temperatures.
However, at elevated temperatures, when studying structural
transitions, phonon entropy plays a much more important role than
electron entropy Bruce . Phonon free energy Fph
can be readily calculated by using density functional perturbation
theory or frozen phonon method
Togo-phonopy2015 ; PhysRevB.71.205214 .
Therefore we approximate Fele(V,T) as the zero-temperature
total energy E0 Togo-phonopy2015 ; PhysRevB.71.205214 . Furthermore
we notice that thermal expansion in solids is usually small and thus
we use the volume of the zero-temperature crystal structures. Thus,
the Helmholtz free energy in Equation (1) is approximated as
[TABLE]
The phonon free energy Fph(T) is defined as
[TABLE]
where Eph(T) is the phonon energy and Sph(T)
is the phonon entropy, both at the volume of zero-temperature crystal structure.
More specifically,
the phonon Helmholtz free energy Fph(T) can be calculated from the phonon frequencies by Togo-phonopy2015
[TABLE]
where q, j, ω, T, and kB are wave vector,
band index of phonon dispersions, phonon frequency, temperature, and
Boltzmann constant, respectively. In our study, the phonon
frequencies are calculated by combining first-principles calculations
with the supercell method and finite displacement method implemented
in Phonopy Togo-phonopy2015 . The dimensions of supercells of
the Pmm2 post-perovskite, Pmm2 perovskite, and Pmn21
perovskite are 4×2×4 (320 atoms), 3×2×3
(180 atoms), and 2×2×2 (160 atoms) of their unit cells,
respectively. The corresponding k-mesh for DFT calculations
of the supercells of Pmm2 post-perovskite, Pmm2 perovskite, and
Pmn21 perovskite are 8×6×6, 5×3×5,
and 8×3×7, respectively.
The obtained Helmholtz free energy F(T)=E0+Fph(T) as a function of temperature (up to 3000 K) is
shown in Supplementary Figure 2. Under 2100 K, the
post-perovskite Pmm2 is the most stable phase. There is a phase
transition between the post-perovskite Pmm2 and the perovskite
Pmn21 around 2100 K, hence the perovskite Pmn21 becomes the most
stable one above 2100 K.
Supplementary Note 5 The DOS of post-perovskite pmm2 structure
Supplementary Figure 3 shows the total density
of states (DOS) and orbital projected densities of states of
post-perovsite Pmm2 BiPbTi2O6.
The DOS at the Fermi level is mainly composed of O-2p,
Bi-6p and Pb-6p states, with very small contributions
from Ti-3d, Bi-6s and Pb-6s states.
Supplementary Note 6 PbTiO3 under bi-axial strain
In experiment, bulk PbTiO3 displays a spontaneous polarization of
about 0.75 C/m2 at 295 K with c/a ratio of 1.063 and space
group of P4mm PhysRevLett.72.3618 ; PhysRevLett.95.177601 .
In this section, we use first-principles calculations (DFT-PBEsol) to
study PbTiO3 under bi-axial strain. In structural optimizations,
we use an energy cutoff of 600 eV and k mesh of
17×17×17. The convergence thresholds of energy and
atomic Hellmann-Feynman forces are 10−9 eV and 10−4 eV Å*-1*,
respectively. We fix the in-plane epitaxial lattice constants (ax=ay=a) and
allow the out-of-plane lattice constant az=c to change. All the
internal atomic coordinates are fully relaxed. We study two different
polarization orientations: if the polarization is along z-axis,
the state is referred to as “out-of-plane polarization”;
if the polarization is either along x-axis or y-axis,
the state is referred to as “in-plane polarization”.
We change the in-plane epitaxial lattice constant a and study the
energy difference between the “out-of-plane polarization” and the
“in-plane polarization” states as a function of a. The results are
shown in Supplementary Figure 4a. There is a critical
lattice constant ac≃3.97 Å at which the two states are
degenerate. If the epitaxial lattice constant is less than ac, the
“out-of-plane polarization” state is more energetically favorable.
If the epitaxial lattice constant exceeds ac, the “in-plane
polarization” state becomes more
stable. Supplementary Figure 4b shows the c/a ratio
of both “out-of-plane polarization” and “in-plane polarization”
states as a function of the in-plane epitaxial lattice constant
a. We note that without any strain, our calculations find that bulk
PbTiO3 has lattice constants ax=ay=3.89 Å and
az=4.17 Å with its polarization pointing along z axis. This
indicates that if we want to stabilize an in-plane polarization in
PbTiO3 thin films, we need at least 2% bi-axial tensile strain.
Supplementary Note 7 Density of states of BiPbTiO6/PbTiO3 interface
Total density of states and
layer-resolved density of states projected onto Ti-3d orbitals for
both parallel and anti-parallel states are shown in
Supplementary Figure 5.
The layer-resolved conduction electrons in the main text are calculated
by integrating the partial density states of Ti-3d orbitals.
Supplementary Note 8 The charge leakage in BiPbTi2O6/ferroelectric heterostructure
We note that in Fig. 5 of our main text charge leakage into PbTiO3
is non-negligible in the BiPbTi2O6/PbTiO3 heterostructure.
This charge leakage is due to “proximate
effect” that PbTiO3 has empty Ti d0 states while BiPbTi2O6
has 0.5e in the d orbitals per Ti site. Charge transfer occurs
from the Ti atoms in BiPbTi2O6 to the Ti atoms in PbTiO3 thin films, a
phenomenon similar to LaTiO3/SrTiO3 interface hwang2002 ; millis2004 .
To prevent charge leakage, we can use PbTi1-xZrxO3 (PZT) to
replace PbTiO3. The mechanism is that Zr has 4d orbitals, whose
energy is higher than Ti 3d orbitals. To demonstrate that, we
replace PbTiO3 by PbZrO3 in our heterostructure and re-do the
calculations. Supplementary Figure 6 shows the conduction electrons in
Ti-d and Zr-d orbitals. We find that charge leakage is completely
suppressed and all the conduction electrons are confined in Ti-d
orbitals in BiPbTi2O6. A direct simulation of PZT requires a much larger
supercell and a proper treatment of random alloying, which is beyond
the capability of our computation resources. However, the physics
trend is clear: the more Ti atoms are replaced by Zr atoms, the weaker
the charge leakage. Usually PZT with x∼0.2−0.5 is widely used in
ferroelectric
heterostructures NatCommPZT-hetero ; ChoiAPL-2010 ; Feigl-JAP2009 ; WangGS-APL2001 .
Supplementary Note 9 Polar displacements in thin films of polar metal BiPbTi2O6
There is “size effects” in ferroelectric thin
films Li_1997JSAP . With the thickness of ferroelectric thin
films decreasing, depolarization fields (if not fully screened) reduce
and sometimes completely suppress the
polarization batra1972thermodynamic ; junquera2003critical .
However, polar metals do not have such a “size effect” because free
electrons in metals fully screen depolarization fields in both bulk
and thin films. To support that, we perform two additional calculations.
The first calculation is a thought-experiment. We calculate a
free-standing one unit cell BiPbTi2O6 thin film. We find that down to one unit
cell, BiPbTi2O6 is still polar. The polar displacements of free-standing
one unit cell BiPbTi2O6 thin film are shown in Supplementary Figure 7, which
are compared to bulk BiPbTi2O6. This shows that there is no “size effect”
in thin films of polar metal BiPbTi2O6.
The second calculation is to study “proximity effect” in BiPbTi2O6 thin
films. Instead of a BiPbTi2O6/PbTiO3 heterostructure, we calculate a
BiPbTi2O6/SrTiO3 heterostructure, in which SrTiO3 is paraelectric.
Supplementary Figure 8 shows the optimized structure in our DFT
calculations and the corresponding polar displacements along x-axis.
We find that the polar displacements in BiPbTi2O6 still exist. In
addition, the structural coupling between BiPbTi2O6 and SrTiO3 drives the
interfacial Ti atom in SrTiO3 to be polar (see
Supplementary Figure 8). This indicates that the polar
displacements in BiPbTi2O6 thin films are not due to proximity
coupling with PbTiO3. In fact, they are so strong that they can
drive a paraelectric material (such as SrTiO3) to be polar close to
the interface.
Supplementary Note 10 Switching of multi-layer BiPbTi2O6 films
To support the physical picture of switching multi-layer BPTO in the main text, we perform
calculations of two-unit-cell BPTO thin films on
PbTiO3. Supplementary Figure 9 shows different configurations
of polar displacements of BPTO and polarization of PbTiO3. The red
arrow is the polarization of entire PbTiO3 thin film. The green
arrow refers to the polar displacements of each unit cell of
BPTO. Since there are two unit cells of BPTO, then there are
altogether four different configurations. Our calculations find that
their total energies are EI<EII<EIII<EIV. This energy order is easy to
understand: both the interface and bulk BPTO prefers to have a
parallel coupling between polar displacements and polarization. In
configuration I, both the polar displacements of BPTO between the two
unit cells and the polarization of PbTiO3 are parallel, which leads
to the lowest total energy. In configuration IV, the polar
displacements of BPTO between the two unit cells are antiparallel.
The polar displacements of the bottom layer BPTO is also
antiparallel to the polarization of PbTiO3.
The two antiparallel couplings combined result in the highest
total energy. The switching process is as follows: we start from
configuration III in which the polarization of PbTiO3 is switched
by an electric field. The interfacial coupling drives the bottom unit
cell of BPTO to switch its polar displacements (i.e., configuration
II). Then the bulk coupling in BPTO drives the top unit cell of
BPTO to switch its polar displacements (i.e., configuration I).
This entire process is favored by thermodynamics because the total
energy monotonically decreases from configuration III to II and
finally to I.
Supplementary Note 11 Uniform strain in epitaxial BiPbTi2O6 thin film from experimental perspective
In our DFT calculations, the BPTO/PbTiO3 heterostructure is
simulated by constraining its in-plane lattice constant to that of
KTaO3 substrate (a = 4.00 Å, see
Supplementary Table LABEL:tab:substrate). The entire heterostructure is under
uniform tensile strain. Under this strain, the Pmm2 perovskite phase
is indeed stabilized (see Fig. 4 in the main text). We find the
metastable (even unstable and unusual polymorphs) phase can appear via
applying uniform epitaxial strains to thin
films trampert1997direct ; XuYaobin-ACS2019 ; PhysRevB.85.024113 ; Sando-APRev2016 .
Such epitaxial stabilization can be understood by the theory of free
energy minimization, in which the energy of coherent and semicoherent
interfaces is much lower than that of noncoherent
ones GorbenkoChemMater2002 . Therefore, the formation of
low-energy interfaces and the minimization of overall free energy of
the system due to the contribution of volume strain energy usually
give rise to the experimentally observed metastable and even unstable
structures.
Specifically for oxide heterostructures, experimentalists find that the critical
thickness below which the entire thin film is under uniform
strain is typically about 10 nm and sometimes can exceed 100
nm Wang2013 . In our case, the thickness of BPTO/PbTiO3
heterostructure (in Fig. 4 in the main text) is only 4 nm, which is
far below the critical thickness for uniform strain.
Furthermore, in our BPTO/PbTiO3 heterostructure, PbTiO3 has a
polarization parallel to the interface. This means that the
depolarization field in PbTiO3 thin film can be fully screened by
the two electrodes (see the “toy device” in
Supplementary Figure 10). Therefore the thickness of PbTiO3
films can be further reduced without suppressing its polarization.
Based on the above reasons, our BPTO/PbTiO3 heterostructure is
anticipated to be uniformly strained and thus can be stabilized on
KTaO3 or NdScO3 substrate.

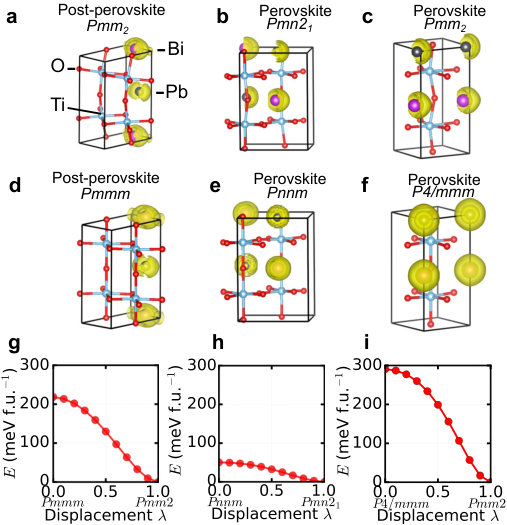 Figure 1
Figure 1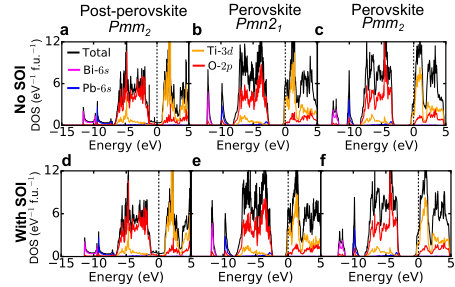 Figure 2
Figure 2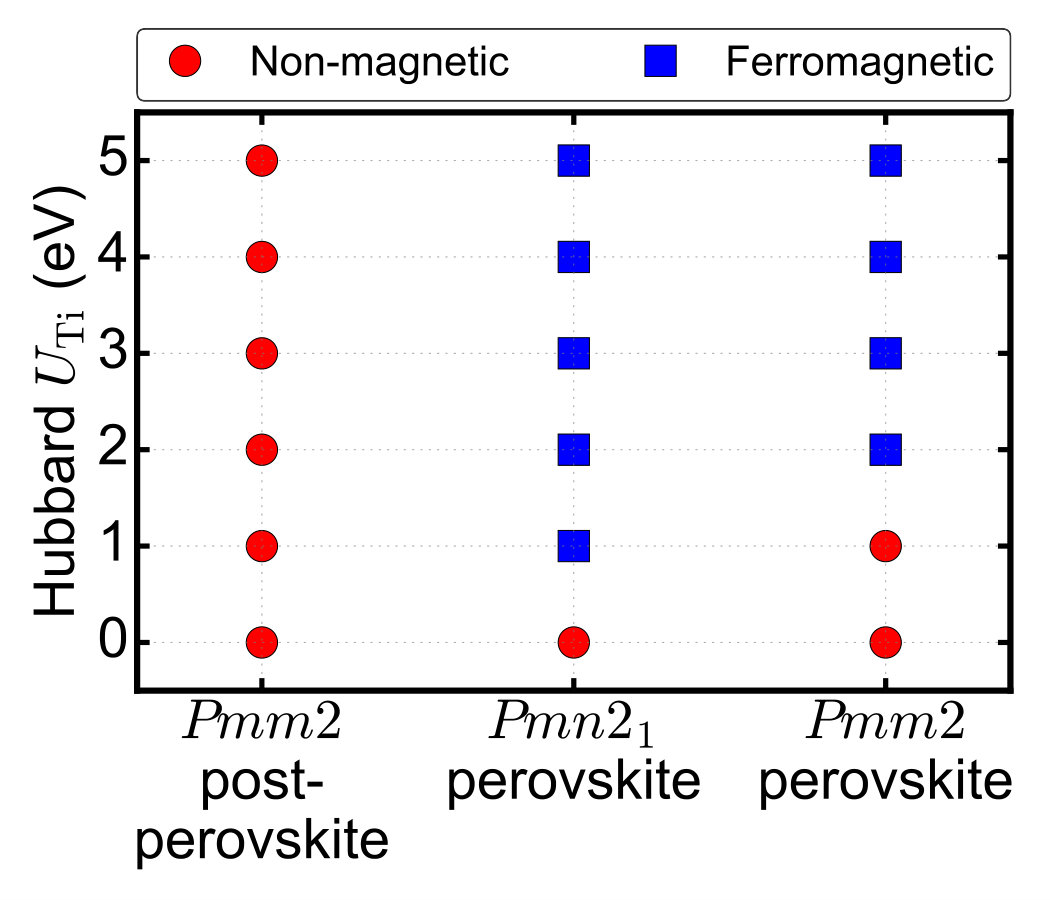 Figure 3
Figure 3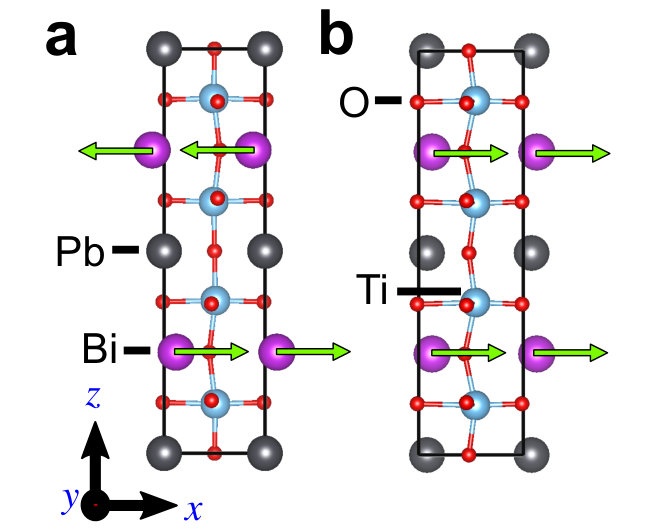 Figure 4
Figure 4 Figure 5
Figure 5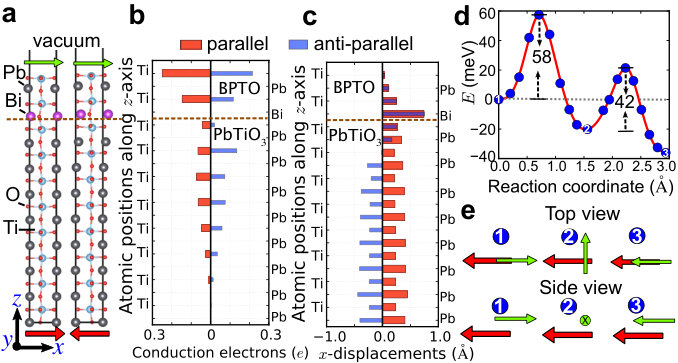 Figure 6
Figure 6 Figure 7
Figure 7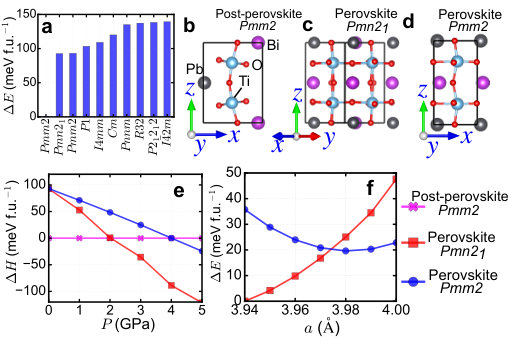 Figure 8
Figure 8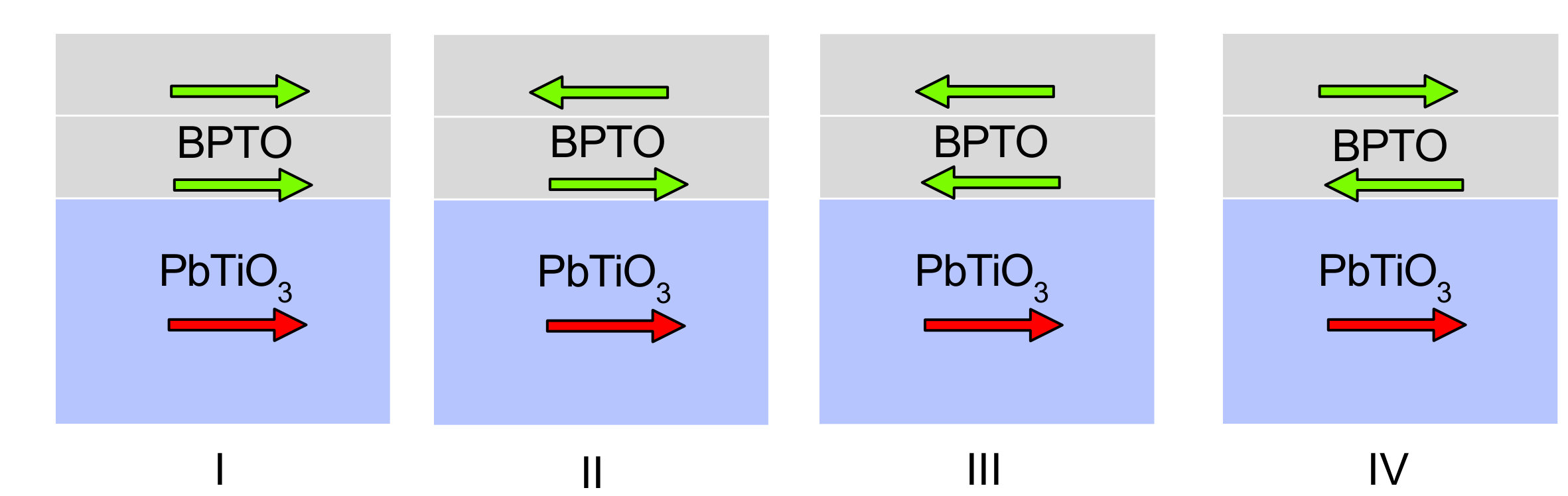 Figure 9
Figure 9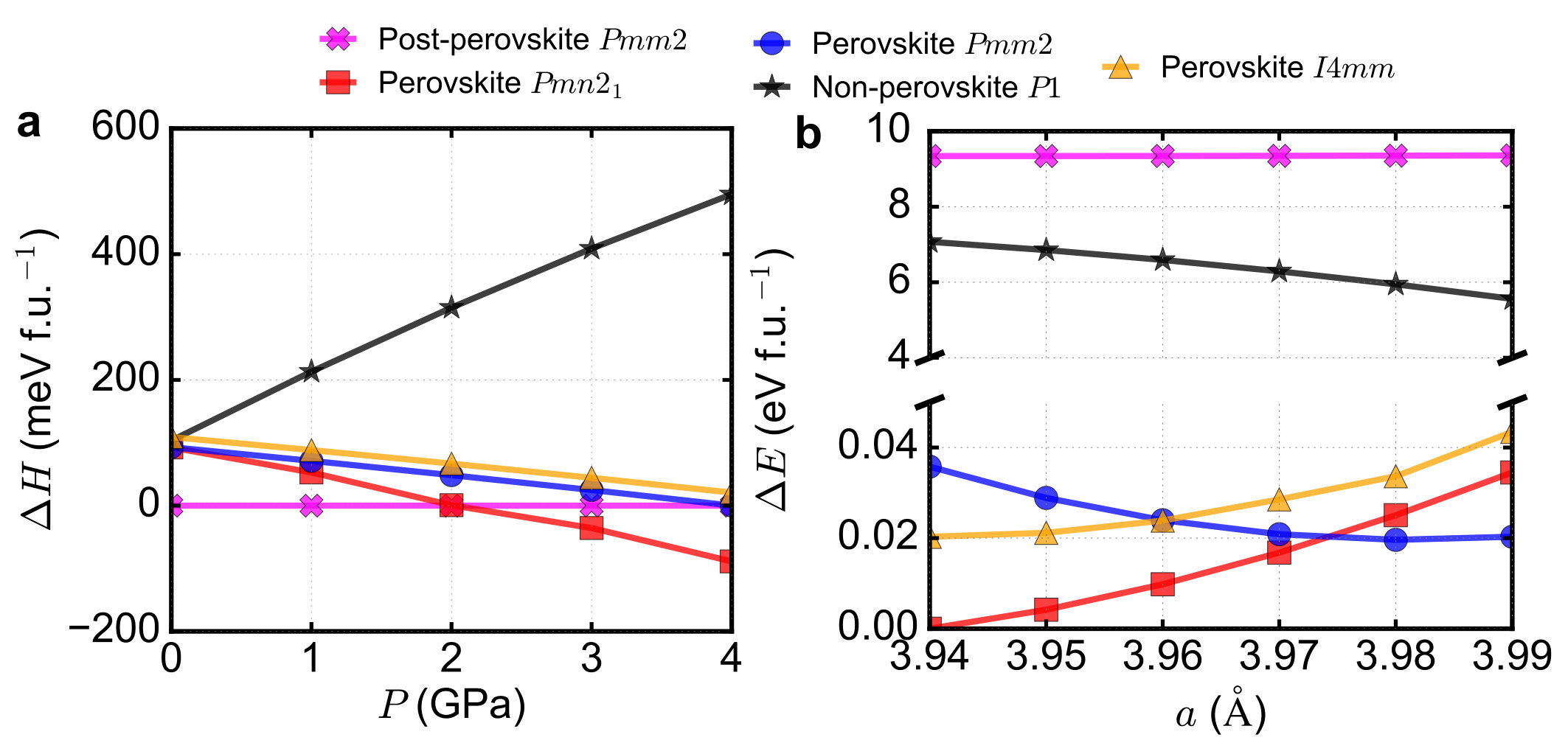 Figure 10
Figure 10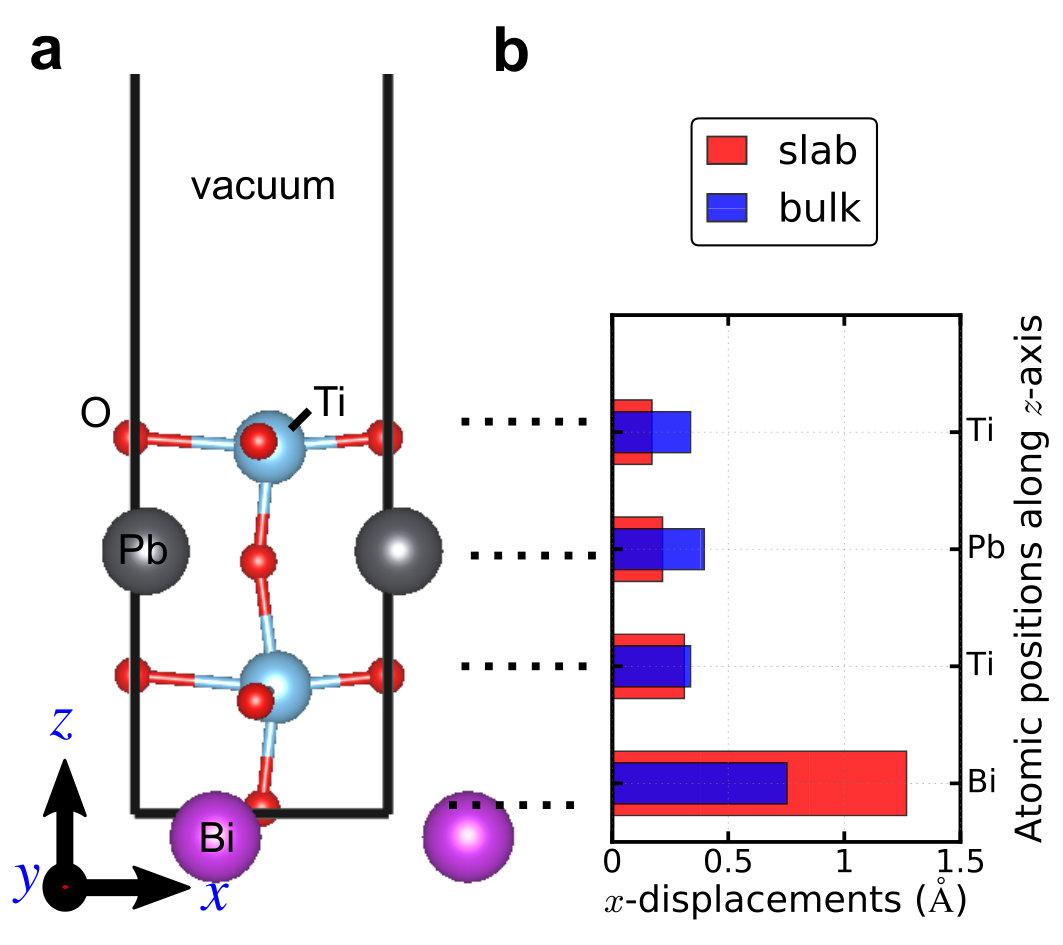 Figure 11
Figure 11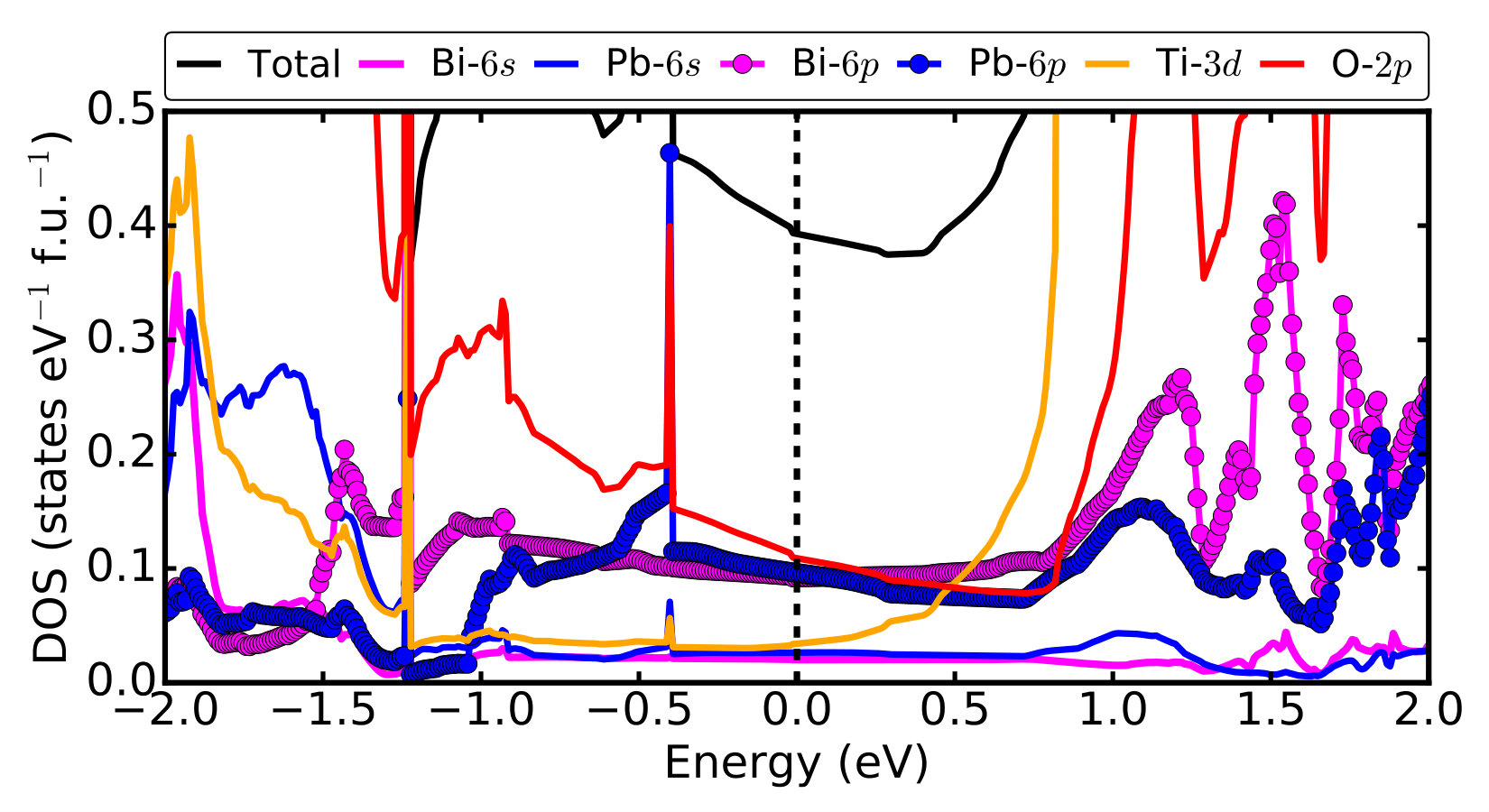 Figure 12
Figure 12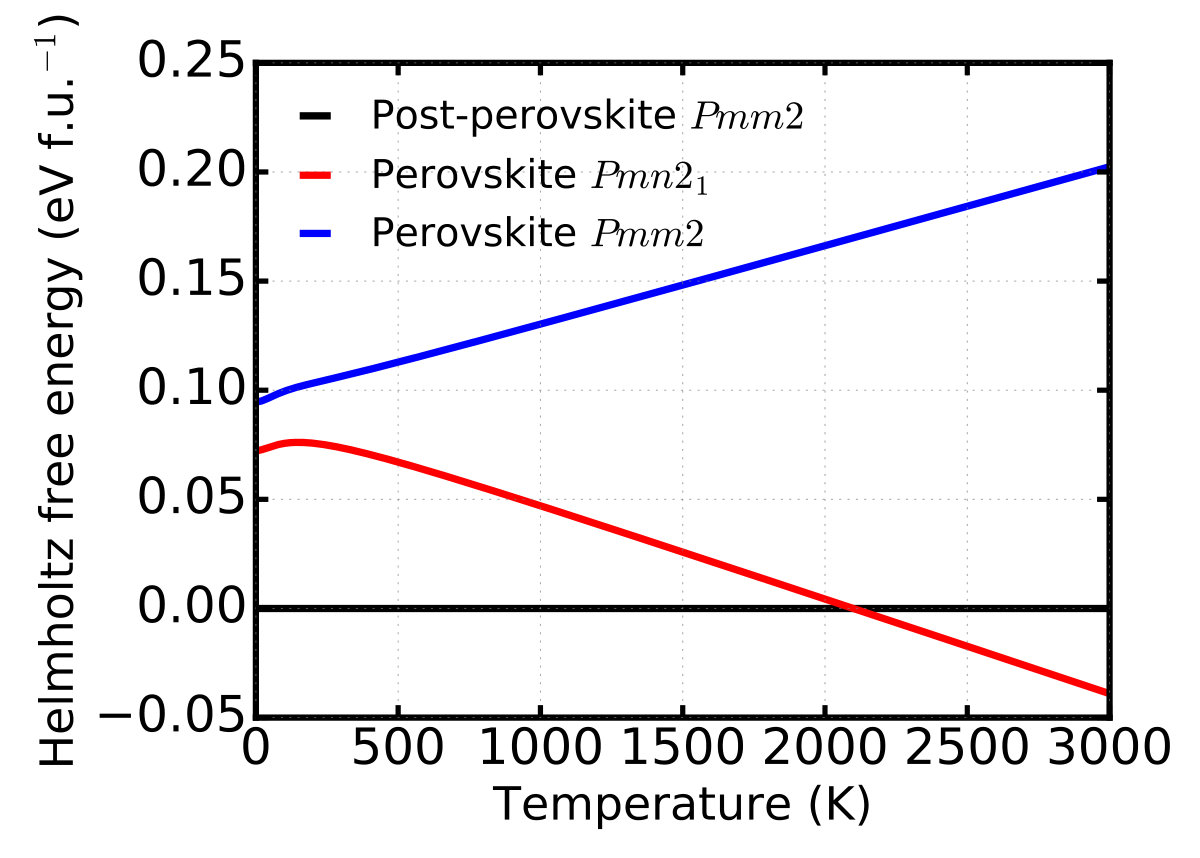 Figure 13
Figure 13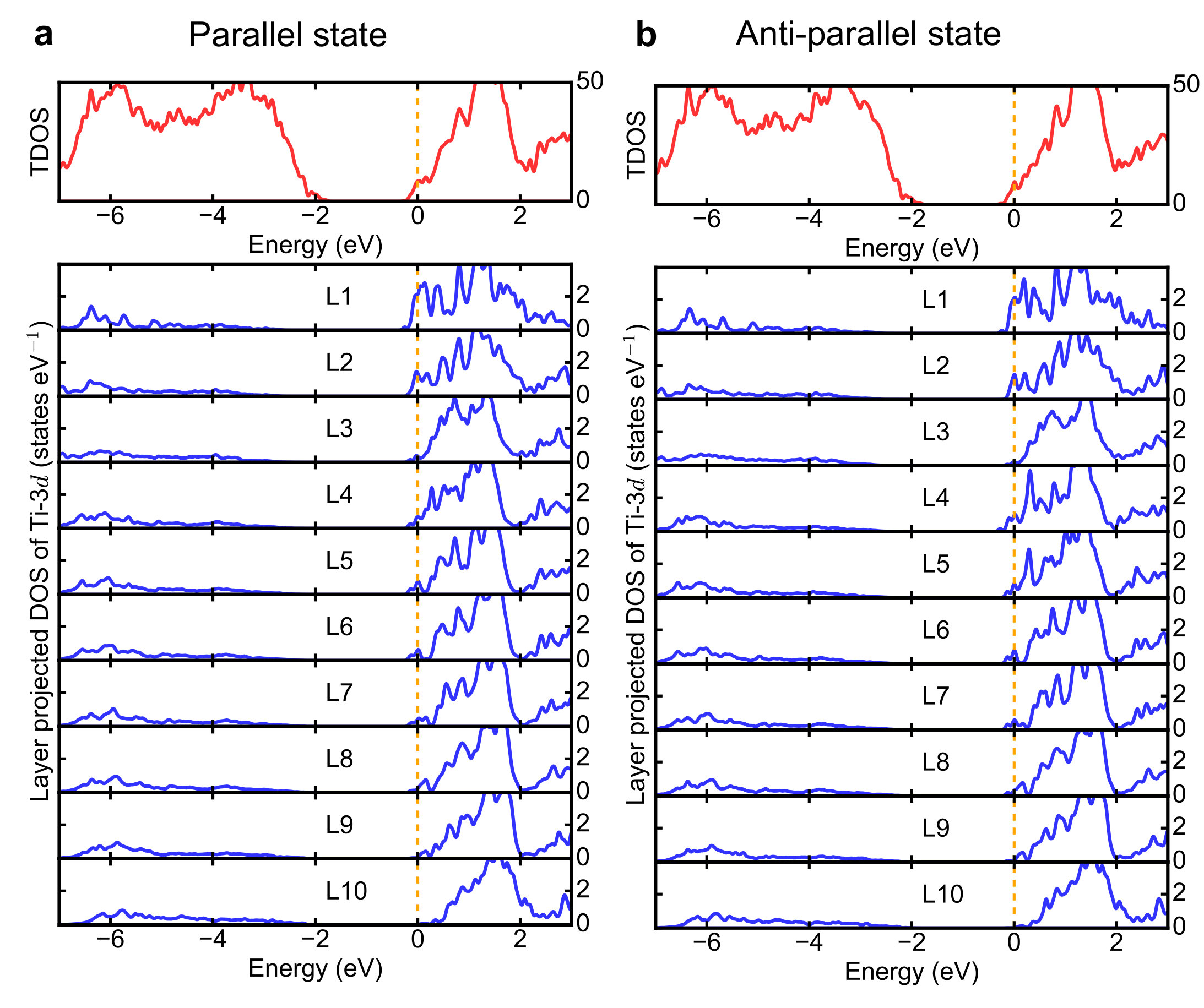 Figure 14
Figure 14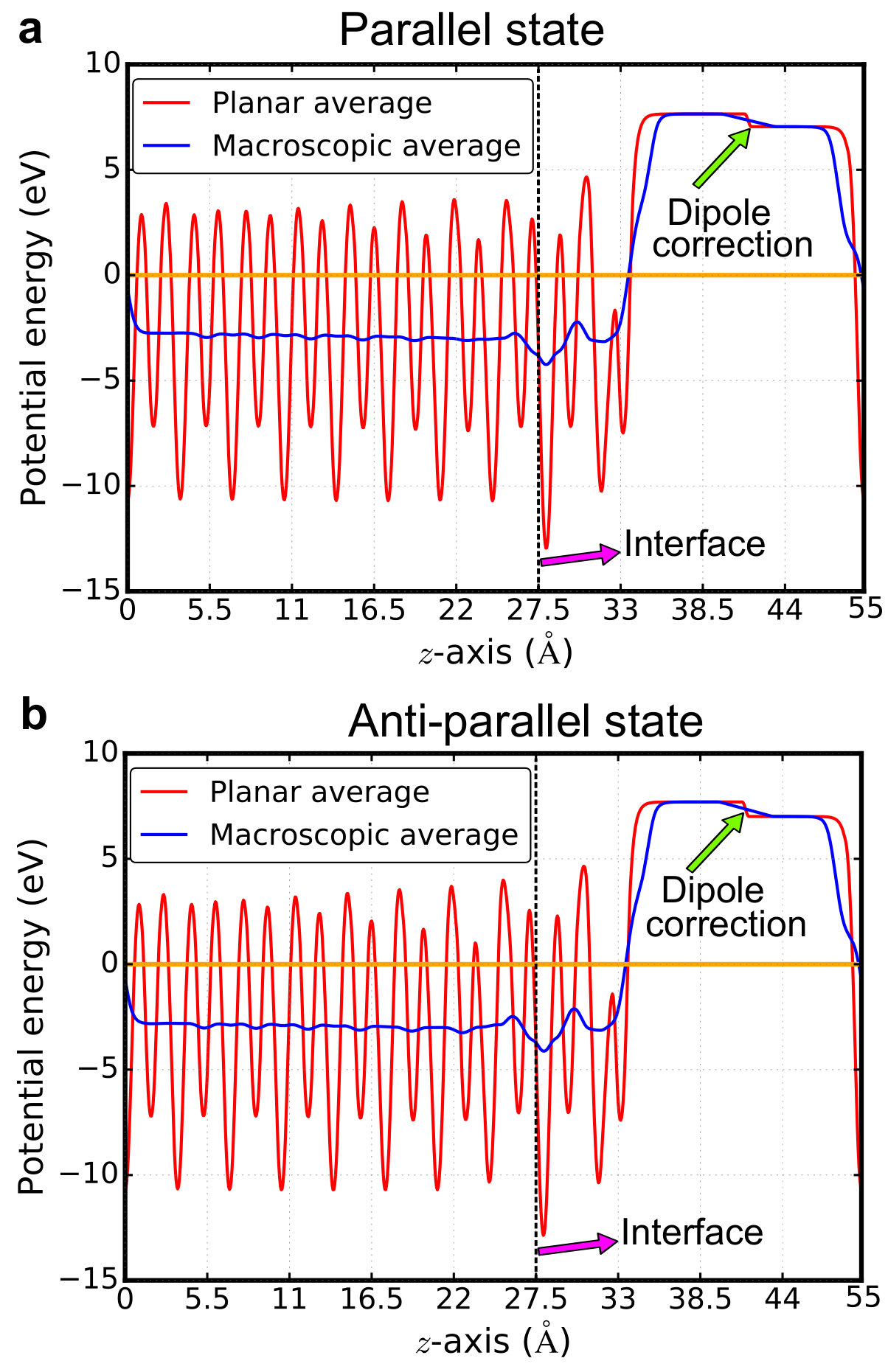 Figure 15
Figure 15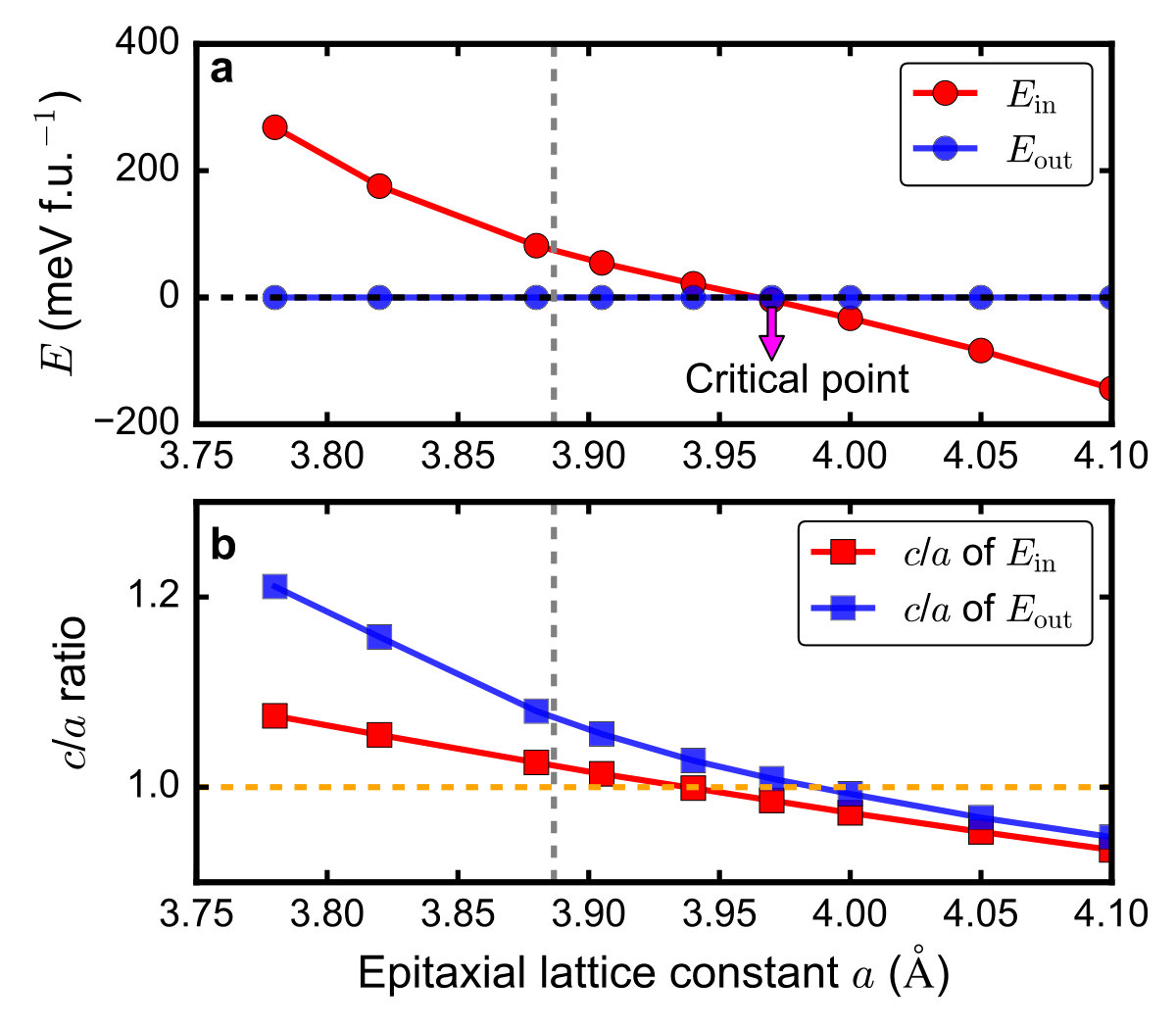 Figure 16
Figure 16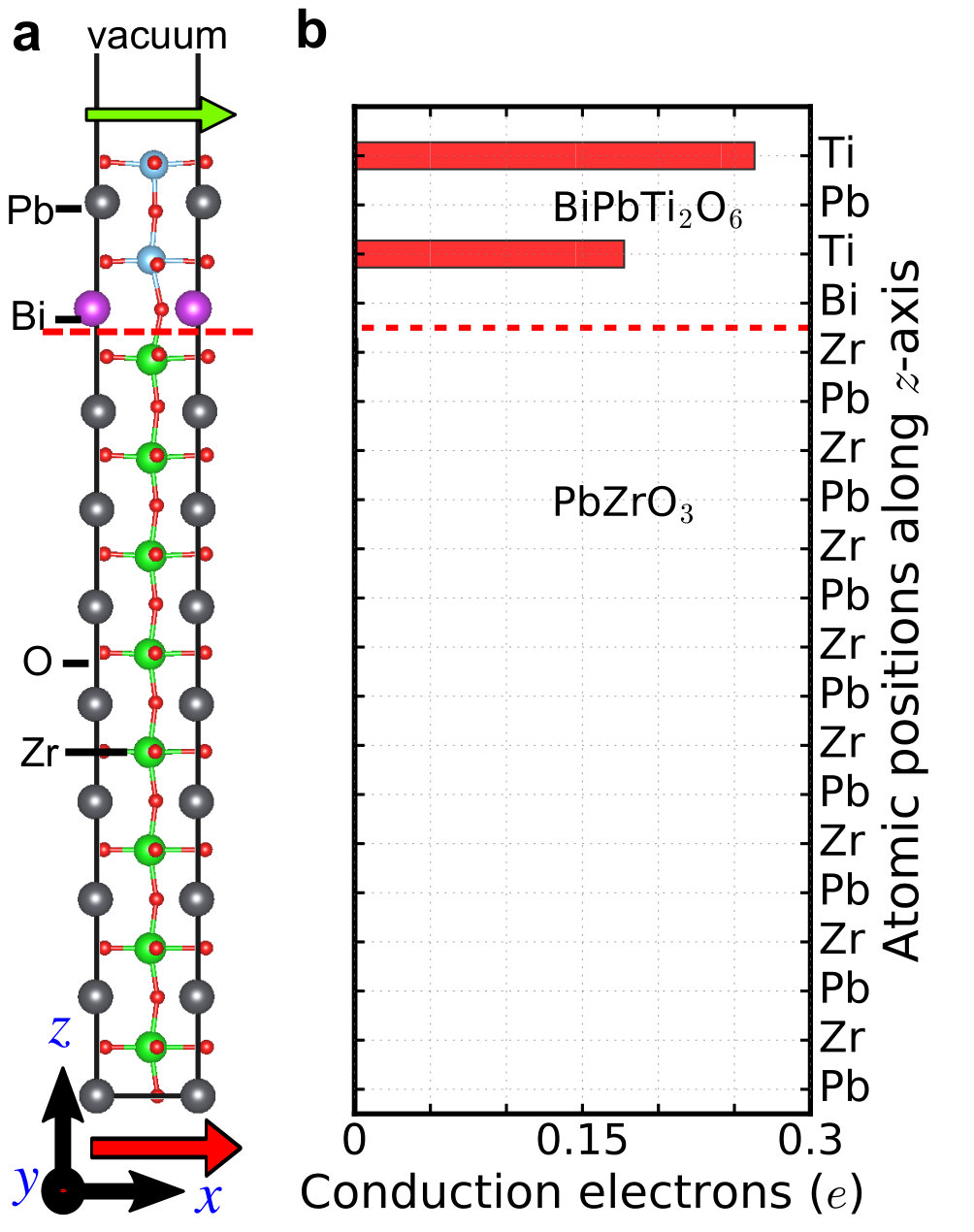 Figure 17
Figure 17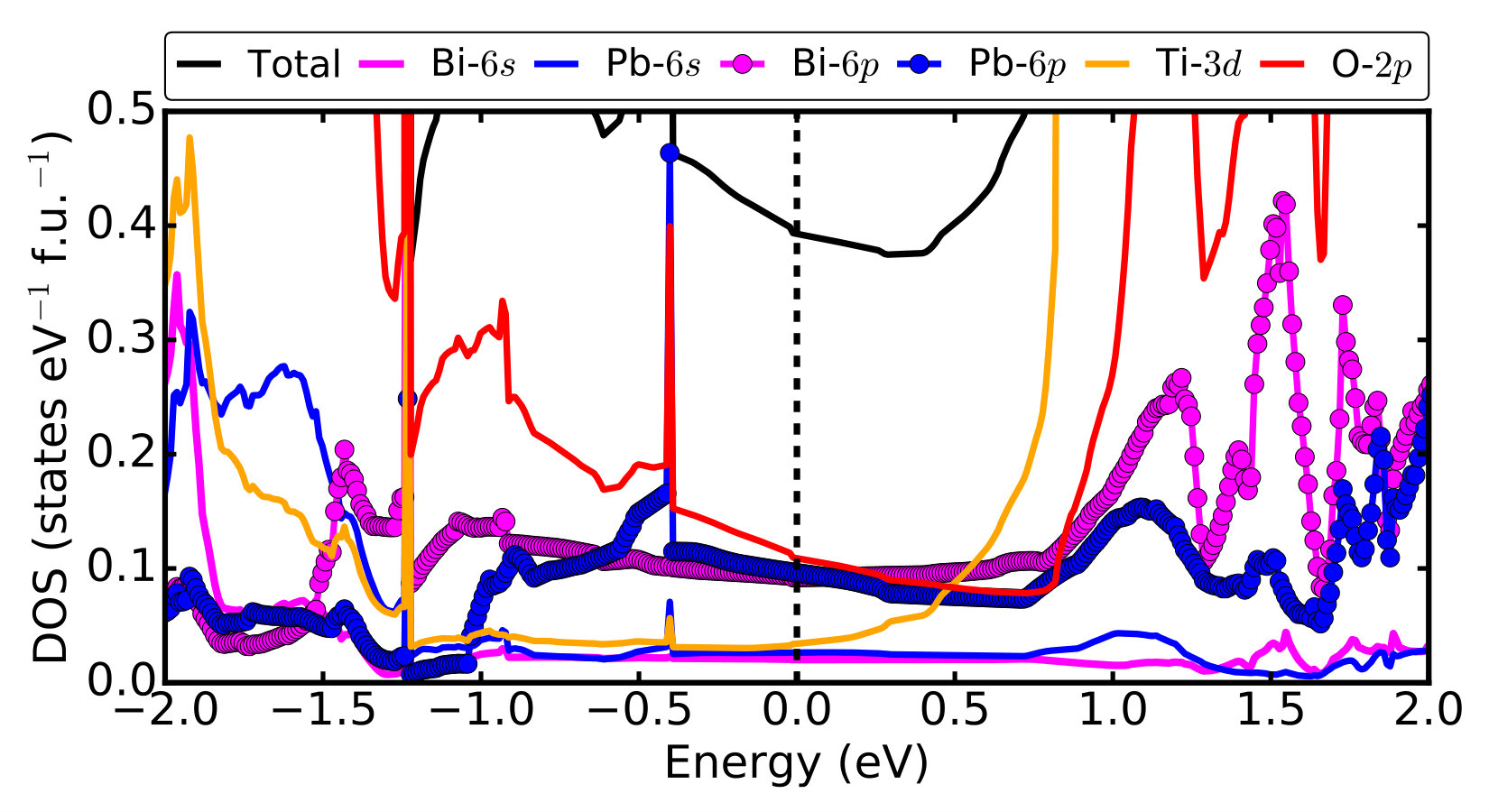 Figure 18
Figure 18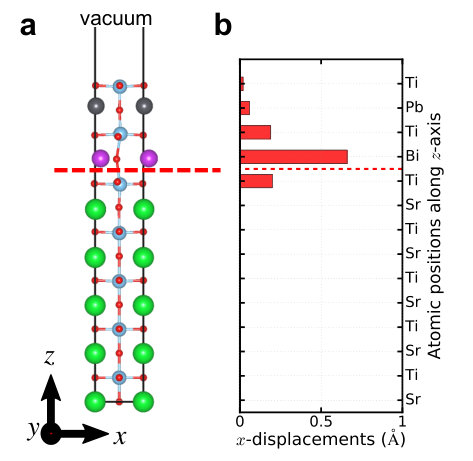 Figure 19
Figure 19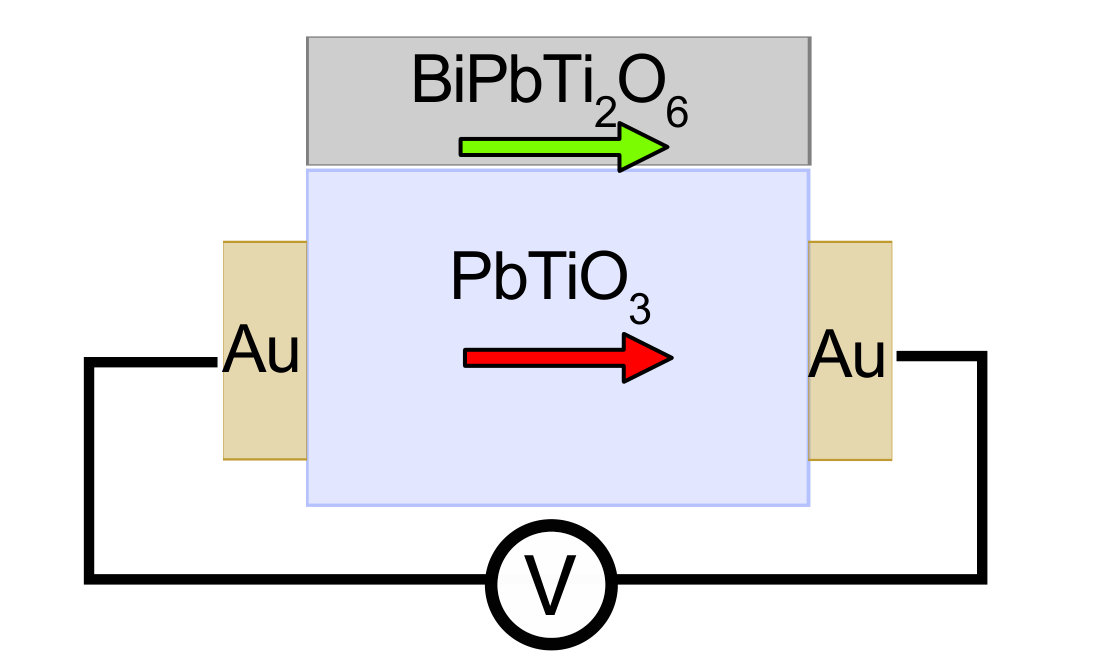 Figure 20
Figure 20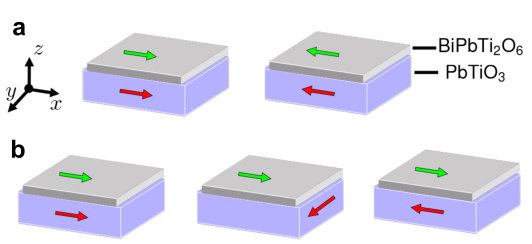 Figure 21
Figure 21