Atomistic Relaxation Process in a Ni3Al Ordered Phase Using Path Probability Method with Vacancy Mechanisms
Ryo Yamada, Tetsuo Mohri

TL;DR
This paper extends the path probability method to explicitly include vacancy mechanisms in the relaxation process of Ni3Al ordered phases, offering a more accurate atomic migration model in alloy systems.
Contribution
It introduces a computational approach that incorporates vacancy mechanisms into the PPM using cluster probabilities, enabling detailed relaxation analysis in alloy phases.
Findings
Vacancy concentration significantly affects relaxation dynamics.
The method successfully models atomic configuration relaxation in Ni3Al.
Explicit vacancy treatment improves the accuracy of atomic migration simulations.
Abstract
The path probability method (PPM), which is a natural extension of the cluster variation method (CVM) to a time domain, has been employed in a relaxation process of atomic configurations in alloy systems. Although the vacancy mechanism is the main atomic migration process in an alloy system, most studies of PPM have used the spin flipping mechanism (or the direct exchange mechanism) because of the huge computational burden imposed by the vacancy mechanism. In this paper the computational problem is circumvented by treating various path variables in the PPM as cluster probabilities in the CVM, and the vacancy mechanism is explicitly taken into account in the theoretical framework. The method is employed to explore the relaxation process in a Ni3Al ordered phase within the tetrahedron approximation, and the effect of vacancy concentration is investigated.
Click any figure to enlarge with its caption.
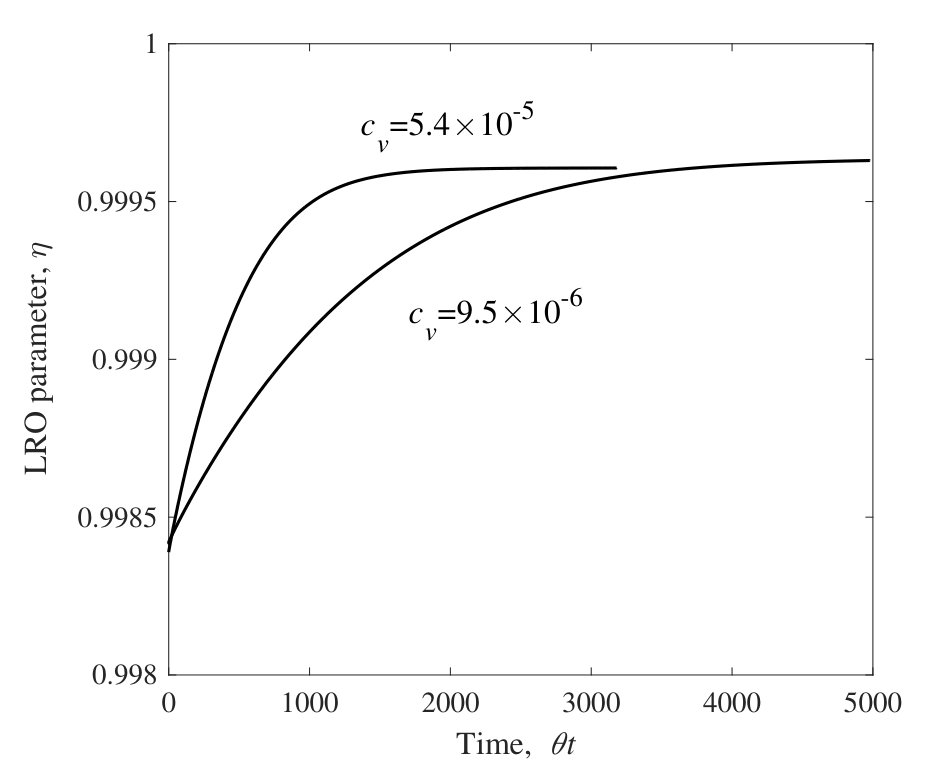 Figure 1
Figure 1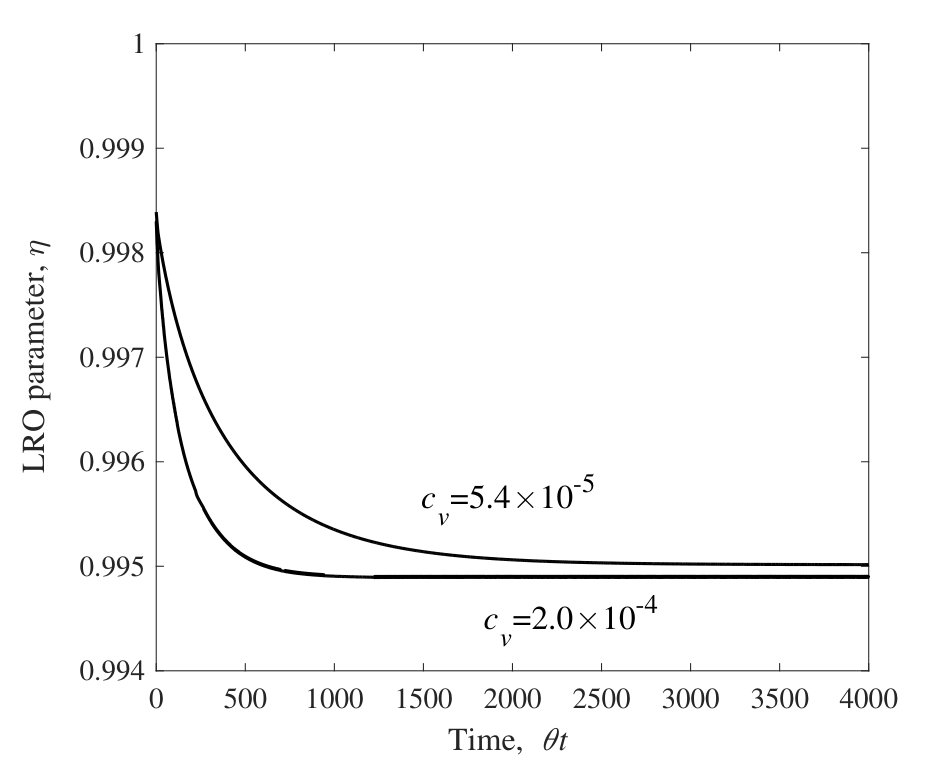 Figure 2
Figure 2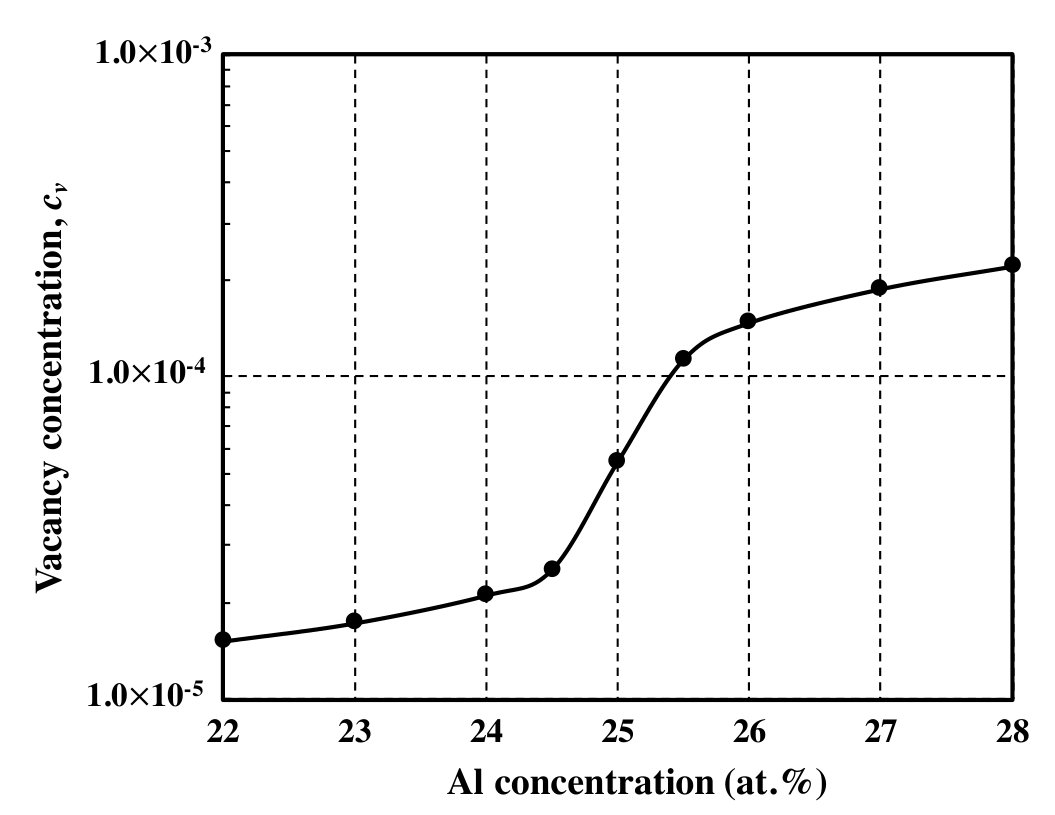 Figure 3
Figure 3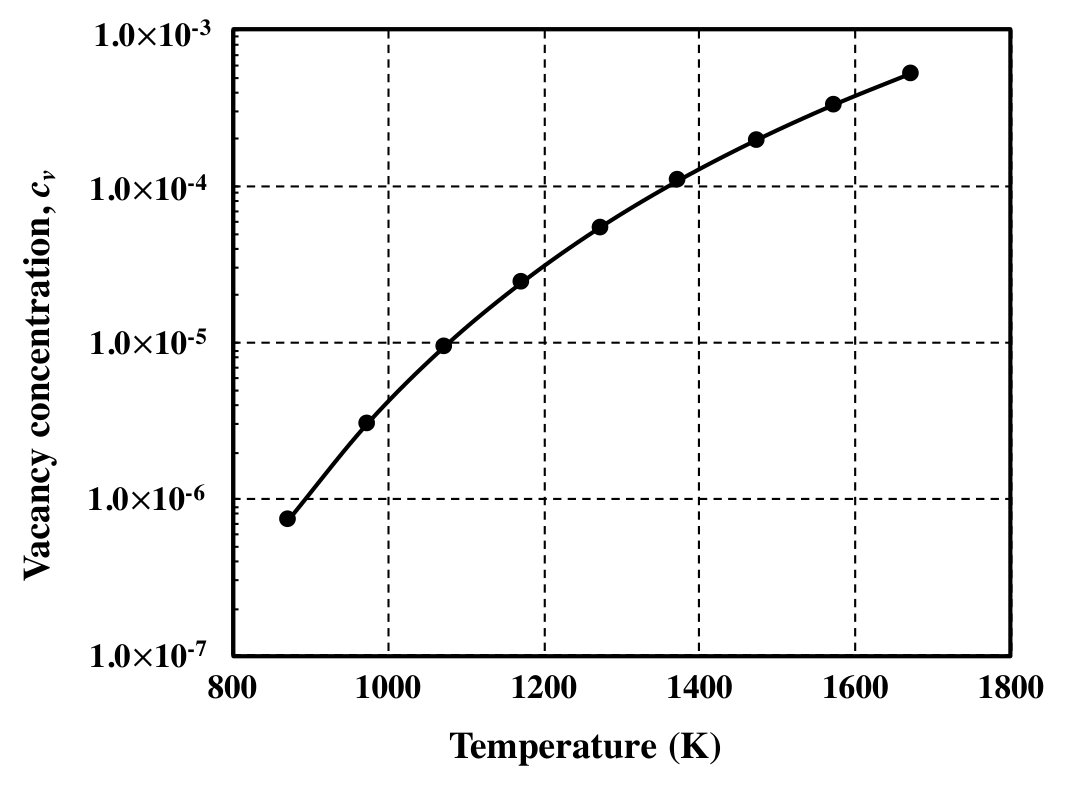 Figure 4
Figure 4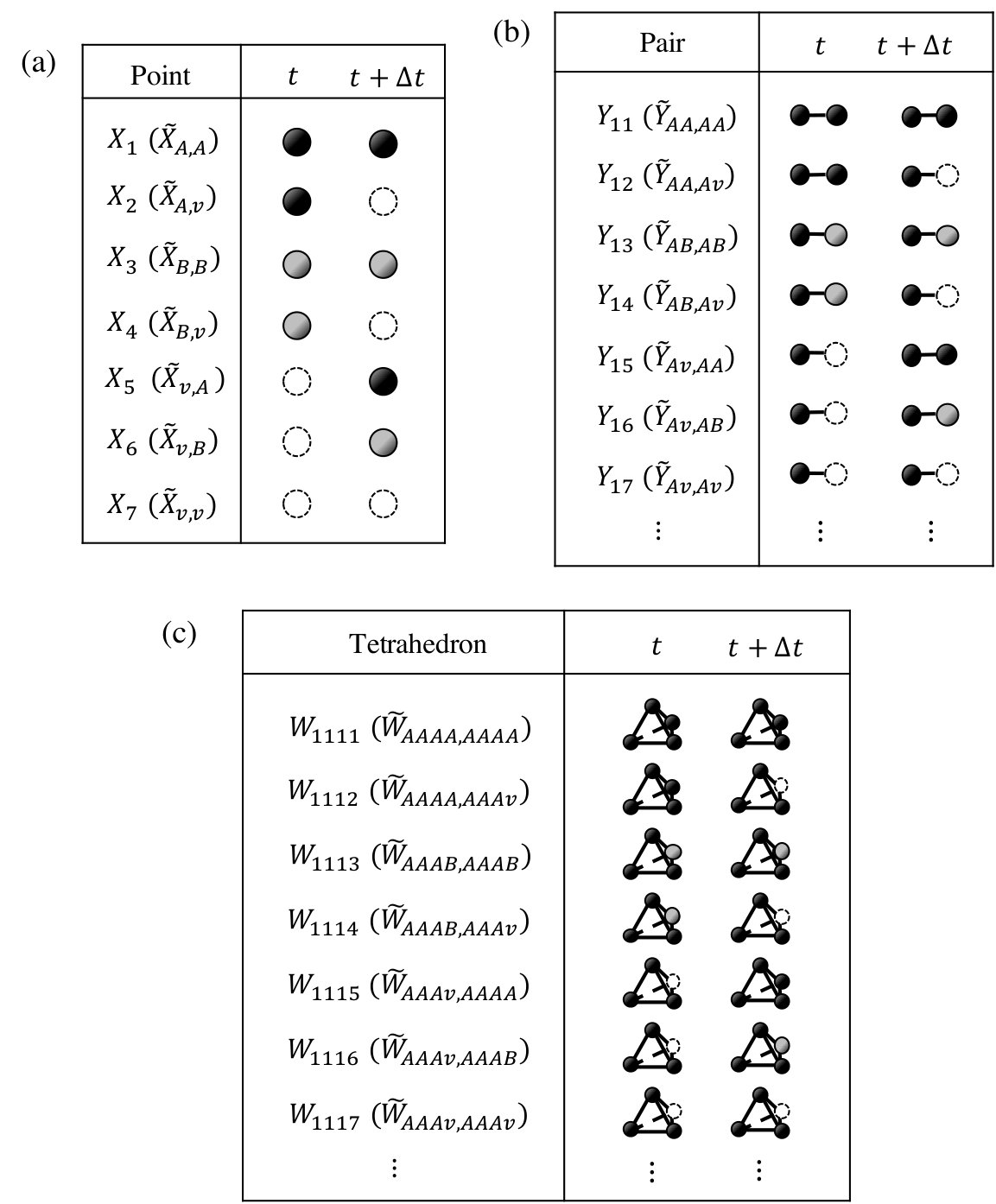 Figure 5
Figure 5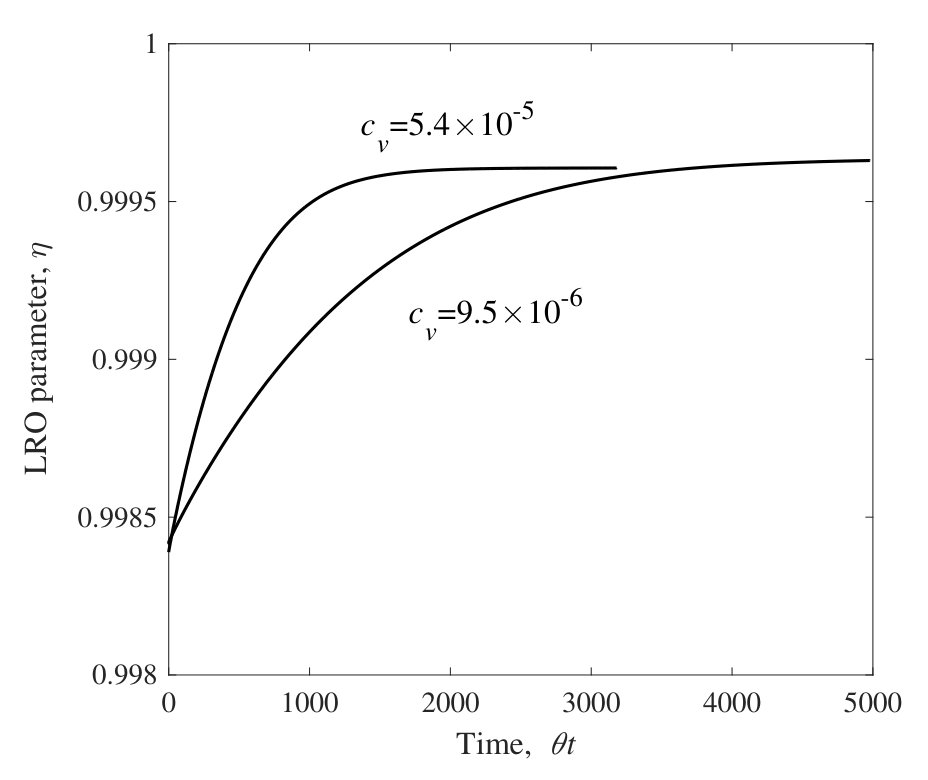 Figure 6
Figure 6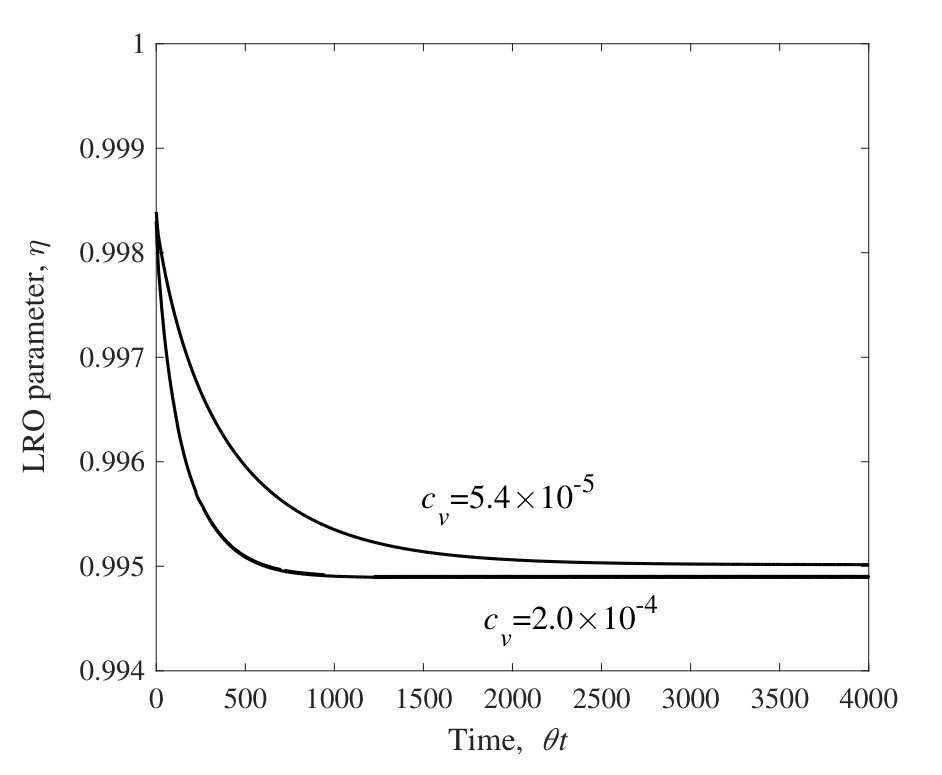 Figure 7
Figure 7 Figure 8
Figure 8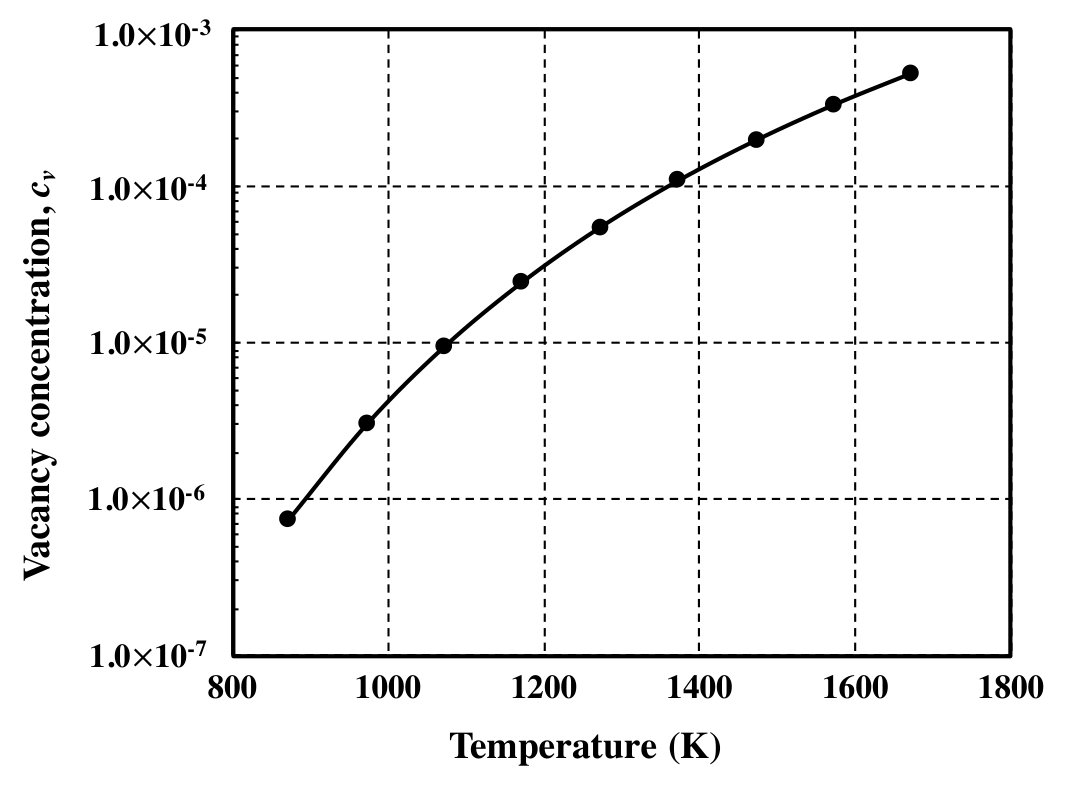 Figure 9
Figure 9 Figure 10
Figure 10| Al–Al | 0.5690 | 2.862 |
| Ni–Ni | 0.7428 | 2.491 |
| Ni–Al | 0.7428 | 2.623 |
| – | – | ||
|---|---|---|---|
| Al– | Al– | ||
| Ni– | Ni– | ||
| –Al | –Al | ||
| –Ni | –Ni | ||
| – | – |
| 1473 K | – | 391.4 | 158.8 |
| 1073 K | 1170.0 | 458.8 | – |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Atomistic Relaxation Process in a Ni3Al Ordered Phase Using Path Probability Method with Vacancy Mechanisms
Ryo Yamada
Institute for Materials Research, Tohoku University, Sendai 980-8577, Japan
Tetsuo Mohri
Institute for Materials Research, Tohoku University, Sendai 980-8577, Japan
Abstract
The path probability method (PPM), which is a natural extension of the cluster variation method (CVM) to a time domain, has been employed in a relaxation process of atomic configurations in alloy systems. Although the vacancy mechanism is the main atomic migration process in an alloy system, most studies of PPM have used the spin flipping mechanism (or the direct exchange mechanism) because of the huge computational burden imposed by the vacancy mechanism. In this paper the computational problem is circumvented by treating various path variables in the PPM as cluster probabilities in the CVM, and the vacancy mechanism is explicitly taken into account in the theoretical framework. The method is employed to explore the relaxation process in a Ni3Al ordered phase within the tetrahedron approximation, and the effect of vacancy concentration is investigated.
pacs:
Valid PACS appear here
I Introduction
Recently, first-principles calculation has received much attention because it enables one to gain some insight into various physical properties which are sometimes difficult to elucidate from experiments. The cluster variation method (CVM) kikuchi1951theory is one of the most reliable theoretical tools for the derivation of configurational entropy, and has been combined with first-principles band structure calculations to determine the properties of materials at finite temperatures mohri1993first ; terakura1987electronic .
Whereas the conventional CVM assumes a rigid lattice of a crystal structure, allowing only a uniform expansion or contraction without changing the symmetry of the lattice, the continuous-displacement cluster variation method (CDCVM) devised by Kikuchi kikuchi1998space incorporates local atomic displacements from a rigid lattice by regarding the displaced atoms as being of a different atomic species located in the Bravais lattice point. This conversion of a degree of freedoms from atomic displacements to atomic configurations is a fruitful approach to the exploration of a displacive transition within the realm of replacive transition. This idea of conversion of freedoms has been applied to various internal freedoms, such as the collective displacements of atoms mohri2013first ; kiyokane2018modelling and spins yamada2019 , leading to structural and magnetic transitions, respectively.
Although an equilibrium state of an alloy system can be reliably determined by the CVM (or CDCVM), one needs another method in order to obtain kinetic information for a system under transition between equilibrium states. The path probability method (PPM) kikuchi1966path , which is a natural extension of the CVM to a time domain, allows one to determine a unique kinetic path without assumptions of near or local equilibrium. The PPM has been applied to order–disorder transitions in binary alloy systems with either a body-centered cubic (bcc) sato1976kinetics or face-centered cubic (fcc) mohri1993kinetic ; mohri1997configurational structure. However, the computational burden is so huge that these applications are still limited to simple model systems. In the calculations for fcc structures, for example, a spin flipping mechanism (or a direct exchange mechanism) is assumed as an atomic migration process, even though a vacancy mechanism is the main mechanism in the atomic diffusion process in alloy systems. The vacancy mechanism was once employed in calculations for bcc alloy systems. However, the pair approximation, which is the simplest approximation in both the CVM and PPM and which demands fewer variables, was used in the special case of equi-atomic alloy composition sato1976kinetics ; gschwend1978kinetics . In order to employ the vacancy mechanism with a higher order approximation for any alloy composition, it would be necessary to explore an efficient computational approach.
In the present work, the basic idea of the CDCVM, that converts additional freedom of local atomic displacements to those of atomic configurations, is extended to PPM calculations, where path variables in the PPM are treated in the same way as cluster probabilities in the CVM. This conversion of freedoms from kinetic paths to configurational ones offers a concise description of kinetic variables in the vacancy mechanism, and the method paves the way to go beyond a simple pair approximation. We demonstrate a successful example in the calculation of relaxation process in a Ni3Al ordered phase, using the tetrahedron approximation.
The organization of this paper is as follows. In the next section, the theoretical backgrounds of the CVM and PPM are briefly described. In the third section, the calculated results are shown and discussed. The final section is devoted to a conclusion.
II Calculation Procedure
II.1 Cluster Variation Method
An initial configuration of the PPM calculations is set up by the CVM. The existence of a vacancy to drive diffusion in an A–B binary system requires a CVM formulation for an A–B– pseudo-ternary alloy system (hereafter, indicates vacancy). To the best knowledge of the authors, there have been only a few attempts at calculation in A–B– alloy systems using the CVM. The work of Shinoda et al. shinoda1992estimation is one of them, where the equilibrium vacancy concentration in a Ni3Al ordered phase is investigated by regarding it as a Ni–Al– pseudo-ternary alloy system. Their calculation procedure is followed here, and some essential points are described below.
A Lennard-Jones type potential is employed to describe the pair interaction energy between two atomic species, and :
[TABLE]
where and are the fitting parameters, and is the interatomic distance. The fitting parameters used in reference shinoda1992estimation are shown in Table 1, and the same parameters are used here.
In order to represent the pair interaction energies for metal–vacancy and vacancy–vacancy pairs, the concept suggested by Doyama and Koehler doyama1976relation is employed. Doyama and Koehler proposed that the metal–vacancy pair interaction energy can be derived by multiplying the “mother” pair energy by 0.35 (this is known as the “ghost bond model” doyama1976relation ; shinoda1995cluster ). In the case of a pure metal, A, the mother pair is A–A, so the metal–vacancy and vacancy–vacancy interaction energies become and , respectively. On the other hand, when a system consists of two different atomic species, A and B, it is difficult to decide which pair is the mother pair for the A–, B–, and – pairs. Fortunately, in the case of ordered phases, one can predict the mother pair from the sub-lattices. Since the Ni3Al ordered phase has a L12 structure, Ni (Al) atoms preferentially exist at the () sub-lattice. Thus, pair interaction energies for metal–vacancy and vacancy–vacancy pairs can be estimated as shown in Table 2.
The tetrahedron approximation is the minimum meaningful approximation that gives a reasonable accuracy and an acceptable computational burden for an fcc-based alloy system mohri2017cluster . Within the tetrahedron approximation, the configurational entropy is given as kikuchi1974superposition ; mohri2013cluster
[TABLE]
where is the total number of lattice points, is the Boltzmann constant, and , , and are cluster probabilities of the point, pair, and tetrahedron, respectively. The superscripts in cluster probabilities indicate sub-lattices.
An equilibrium state of a system is determined from the following conditions:
[TABLE]
[TABLE]
and
[TABLE]
where is the grand potential and is the effective chemical potential for vacancy. The grand potential, , is given as , where is the Helmholtz free energy and is the effective chemical potential for atomic species, . The minimization of the grand potential in terms of cluster probability (Eq. (3)) is conducted by the natural iteration method (NIM) kikuchi1974superposition . Eqs. (3) and (4) are general conditions for the determination of equilibrium states, but Eq. (5) is an additional condition for systems containing vacancies, in which is introduced because the free energy should have a minimum in terms of the number of vacancies under the condition that the number of species, A and B (or Al and Ni), is fixed. The detail of the derivation of Eq. (5) is not discussed here. Readers interested in the derivation should refer to the original paper shinoda1992estimation .
II.2 Path Probability Method
Whereas the minimization of free energy (or grand potential) is required for a determination of equilibrium states in the CVM, a path probability function (PPF), which is a probability of possible paths in a transition process, is maximized in the PPM to determine the most probable path. Before going into details of the PPF, path variables are defined below, which correspond to cluster probabilities in the CVM.
Some representative path variables defined in the present work are shown in Table 3. It should be noted that the vacancy mechanism is reflected in these path variables, and a direct exchange mechanism, such as A to B (or Al to Ni), is not considered. The basic idea of the CDCVM is used in defining these path variables. In the CDCVM, quasi-lattice points are introduced around each Bravais lattice point and an atom displaced into one of these points is regarded as a different kind of atomic species, which locates at the Bravais lattice point. Hence, the freedom of the atomic displacement is replaced by a configurational freedom of a multicomponent alloy. In the path variables shown in Table 3, the path variables for point clusters are treated in the same way as cluster probabilities in the CVM for seven-component systems, suggesting that kinetic freedoms are converted into configurational ones.
The conventional expression for path variables is also shown in Table 3; e.g., describes the transition of tetrahedron configuration --- to --- in . Using this conventional expression, the time evolution/devolution of tetrahedron cluster probabilities are given as
[TABLE]
In order to avoid complicacies of notation, a simplified expression for path variables, , , and , is used throughout this study. The path variables satisfy the following normalization and geometrical conditions:
[TABLE]
and
[TABLE]
The PPF is maximized using NIM as in reference ohno2005iteration , where the relation between the cluster probabilities and the path variables, Eq. (6), and the normalization condition, Eq. (7), are taken into account. The use of the NIM dramatically reduces the computational burden in the PPM.
The PPF is defined as the product of three terms: a probability of an atomic jump, ; a probability of thermal activation for a state change, ; and the number of equivalent paths, . Each term is written as
[TABLE]
[TABLE]
and
[TABLE]
where is the jump probability, is the time step, , , and are path variables of point, pair, and tetrahedron clusters, respectively, is a change of internal energy before and after atomic jumps in , and is the temperature. The jump frequency, , and internal energy change, , are written as follows:
[TABLE]
and
[TABLE]
where is an attempt frequency, is an activation energy, and and are the energy changes of pair interaction energies and the chemical potentials in , respectively. Note that Eq. (2) and Eq. (11) have quite similar forms because Eq. (11) is derived in the same way as the configurational entropy in the CVM. Therefore, Eq. (11) is also called a tetrahedron approximation as well as Eq. (2) in the CVM.
As mentioned above, the maximization of the PPF is conducted by the NIM as well as the minimization of grand potential in the CVM. Using this approach, a relaxation process of atomic configurations in a Ni3Al ordered phase is examined when it is quenched from 1273 K to 1073 K and 1473 K. These relaxation processes are evaluated by the time dependence of long-range order (LRO) parameters, , and their relaxation time, , which are, respectively, defined as
[TABLE]
and
[TABLE]
where and are, respectively, the LRO parameters at the equilibrium and initial states.
III Results and Discussion
III.1 Equilibrium state
The Al concentration dependence of vacancy concentration at 1273 K calculated by the CVM is shown in Fig. 1. It shows a step-like dependence and the order of the vacancy concentration is . These correspond to the results seen in the preceding work shinoda1992estimation . The step-like dependence of vacancy concentration is attributed to the different Al concentration dependences at the two sub-lattices. This interesting behavior strongly depends on the pair interaction parameters used in the calculation (details can be found in reference shinoda1992estimation ).
Additionally, the temperature dependence of vacancy concentration at 25.0 at.% Al is shown in Fig. 2. It can be seen that the vacancy concentration increases with temperature. This is due to the contribution of configurational entropy at high temperatures. These atomic configurations (or cluster probabilities) determined from the CVM are used as an initial state in the PPM calculations (Sec. III.2).
III.2 Kinetic path to the equilibrium state
The time evolution of LRO parameters, , at 25.0 at.% Al during the isothermal aging process at 1473 K and 1073 K are shown in Figs. 3 and 4, respectively. The horizontal lines are normalized by the jump probability, . For these calculations the concentration of vacancy is fixed at the equilibrium value of an initial state (i.e., at 1273 K) or at the value at the isothermal aging temperature (i.e., at 1473 K or 1073 K). Figures 3 and 4 show that the LRO parameters become smaller (larger) with time for the aging at 1473 K (1073 K). The long time limits of their values are confirmed to be the equilibrium ones independently calculated for each temperature by the CVM (see Sec. III.1).
From the results of the LRO parameter, the relaxation time, , is obtained by fitting into Eq. (15). The relaxation times derived from aging at 1473 K and 1073 K are shown in Table 4. From these results, it is confirmed that the more (fewer) vacancies exist, the smaller (larger) the relaxation time the system has. If the vacancy concentrations are allowed to alter during aging, it is expected that the relaxation time would be at some value between the values obtained here.
Note that the horizontal lines in Figs. 3 and 4 are normalized by the jump frequency, . The real time dependence of LRO parameters are determined if the attempt frequency, , and the activation energy, , are known. These values would be calculated from a band structure calculation, but this is beyond the scope of the present study.
IV Conclusions
An idea of conversion of freedoms used in CDCVM is applied to the PPM framework, where kinetic freedoms are converted into configurational freedoms, and the relaxation process in a Ni3Al ordered phase is explored by taking into account the vacancy mechanism as an atomic migration process. It is confirmed that a higher (lower) vacancy concentration in the system leads to a faster (slower) relaxation time.
It is noted that in the present work the timescale of the calculated results is normalized by the jump probability. The jump probability is related to the activation energy and attempt frequency. The incorporation of realistic values can be achieved by combining it with electronic structure calculations, which will be reported elsewhere.
ACKNOWLEDGEMENT
This study is partly supported by a project (P16010) commissioned by the New Energy and Industrial Technology Development Organization (NEDO), and by the Structural Materials for Innovation of the Cross ministerial Strategic Innovation Promotion Program (SIP) of Japan Science and Technology (JST). One of the authors (TM) appreciate their supports. Also, we express sincere appreciation to Dr. Y. Yamada at Materials and Energy Division, CSRC, Beijing for his stimulating discussions.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1(1) R. Kikuchi, “A theory of cooperative phenomena,” Physical review , vol. 81, no. 6, pp. 988–1003, 1951.
- 2(2) T. Mohri, S. Takizawa, and K. Terakura, “First-principles calculation of heats of formation for Au–Cu, Au–Pd and Au–Ag alloys with thermal vibration effects,” Journal of Physics: Condensed Matter , vol. 5, no. 10, p. 1473, 1993.
- 3(3) K. Terakura, T. Oguchi, T. Mohri, and K. Watanabe, “Electronic theory of the alloy phase stability of Cu–Ag, Cu–Au, and Ag–Au systems,” Physical Review B , vol. 35, no. 5, pp. 2169–2173, 1987.
- 4(4) R. Kikuchi, “Space is continuous–continuous-displacement treatment of phase-separating diagrams,” Journal of phase equilibria , vol. 19, no. 5, pp. 412–421, 1998.
- 5(5) T. Mohri, Y. Chen, and N. Kiyokane, “First-principles cluster variation calculations of tetragonal-cubic transition in Zr O 2 ,” Journal of Alloys and Compounds , vol. 577, pp. S 123–S 126, 2013.
- 6(6) N. Kiyokane and T. Mohri, “Modelling of a displacive transformation in two-dimensional system within four-body approximation of Continuous Displacement Cluster Variation Method,” Philosophical Magazine , vol. 98, no. 11, pp. 1005–1017, 2018.
- 7(7) R. Yamada and T. Mohri, “Conversion of magnetic freedoms into atomic configurational freedoms within the Cluster Variation Method,” in preparation .
- 8(8) R. Kikuchi, “The path probability method,” Progress of Theoretical Physics Supplement , vol. 35, pp. 1–64, 1966.