Quasicrystalline electronic states in 30$^\circ$ rotated twisted bilayer graphene
Pilkyung Moon, Mikito Koshino, Young-Woo Son

TL;DR
This paper develops a theoretical framework to describe the electronic states in 30° twisted bilayer graphene, revealing quasicrystalline properties, resonant states, and flat bands with fractal wave function patterns.
Contribution
It introduces a symmetry-respecting theoretical model for 30° twisted bilayer graphene, capturing its quasicrystalline electronic states and resonant phenomena.
Findings
Identification of 12-fold quantized angular momentum states
Presence of nearly flat bands with fractal wave functions
Spatial localization of resonant states in finite samples
Abstract
The recently realized bilayer graphene system with a twist angle of offers a new type of quasicrystal which unites the dodecagonal quasicrystalline nature and graphene's relativistic properties. Here, we introduce a concise theoretical framework that fully respects both the dodecagonal rotational symmetry and the massless Dirac nature, to describe the electronic states of the system. We find that the electronic spectrum consists of resonant states labeled by 12-fold quantized angular momentum, together with the extended relativistic states. The resulting quasi-band structure is composed of the nearly flat bands with spiky peaks in the density of states, where the wave functions exhibit characteristic patterns which fit to the fractal inflations of the quasicrystal tiling. We also demonstrate that the 12-fold resonant states appear as spatially-localized states in a…
Click any figure to enlarge with its caption.
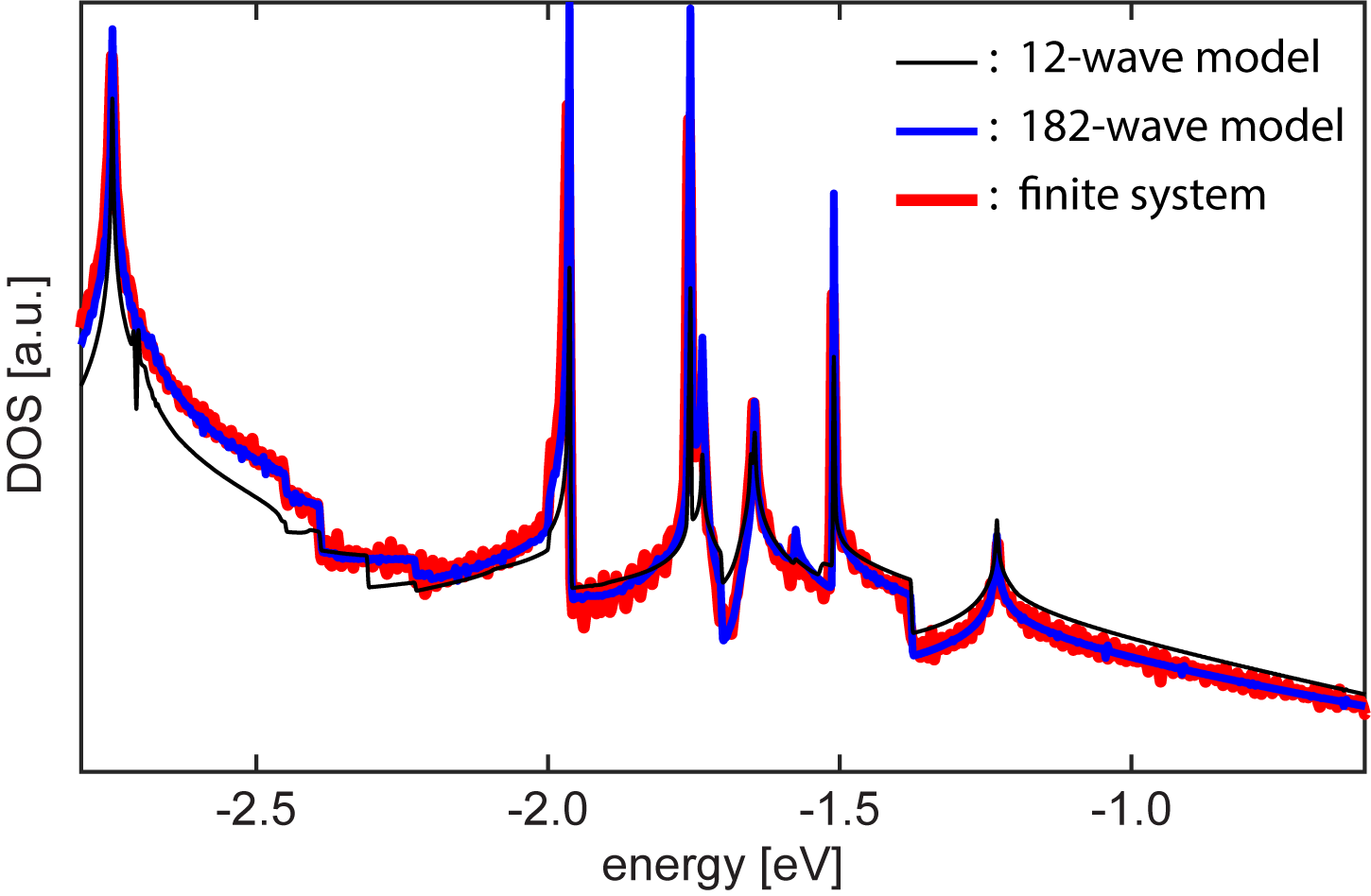 Figure 1
Figure 1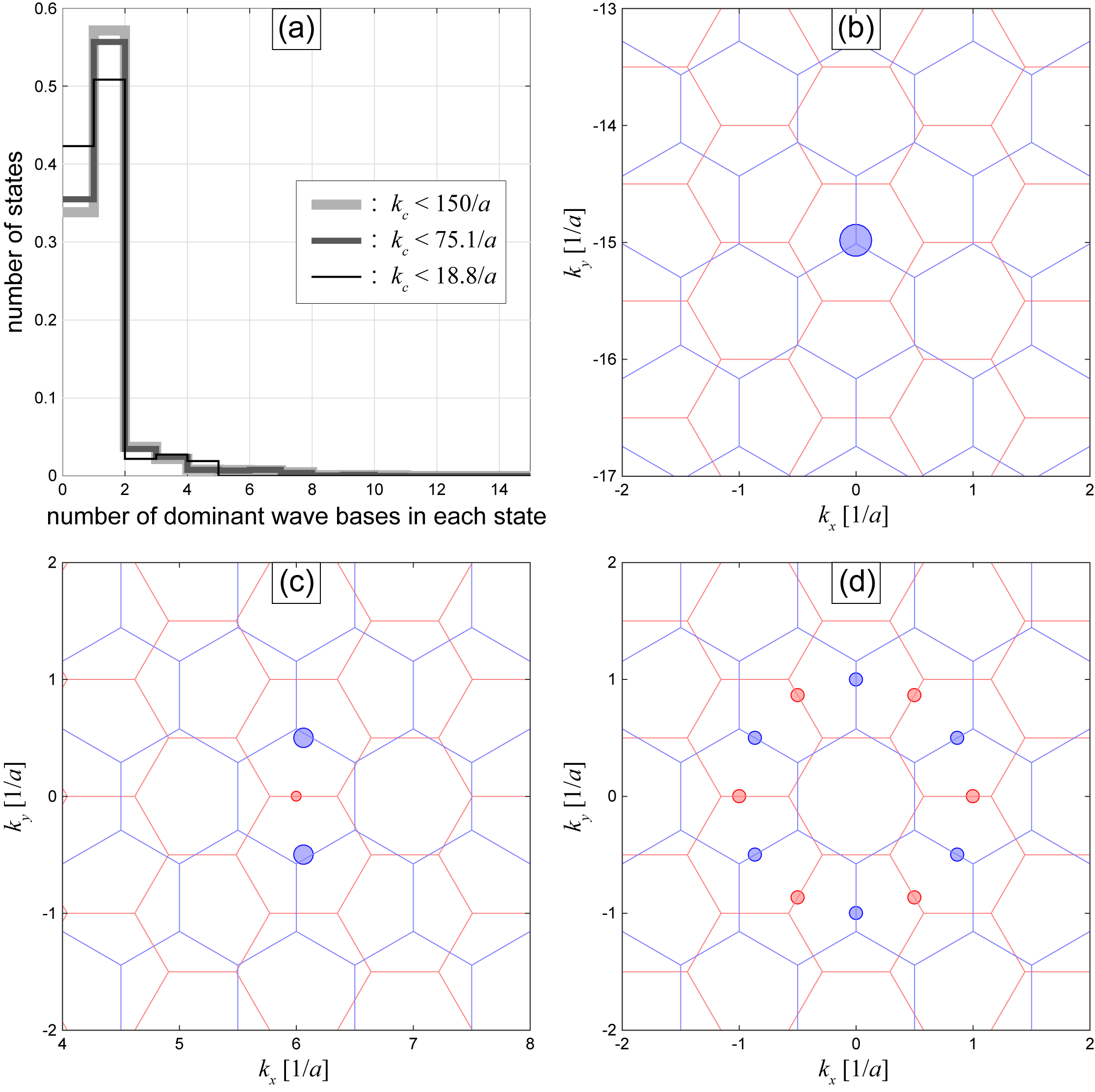 Figure 2
Figure 2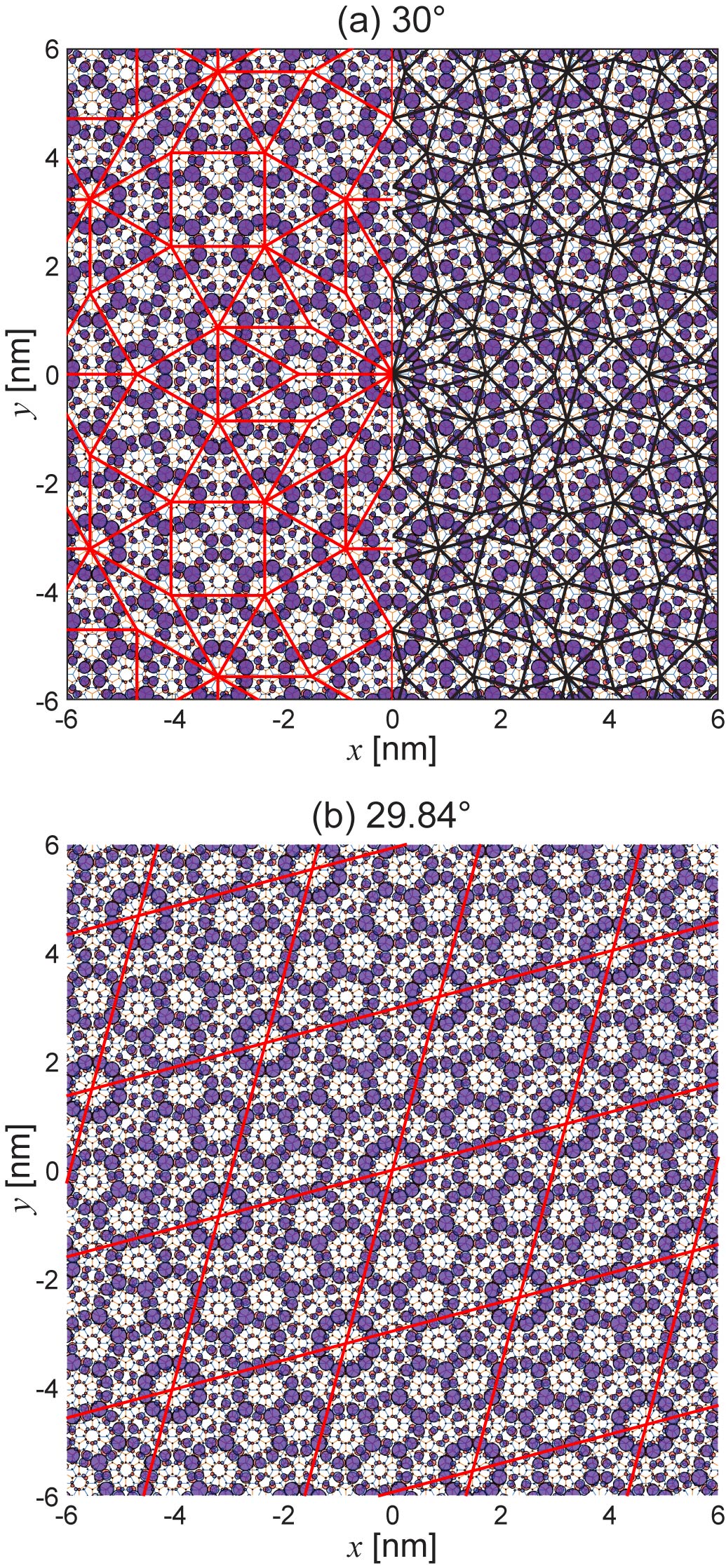 Figure 3
Figure 3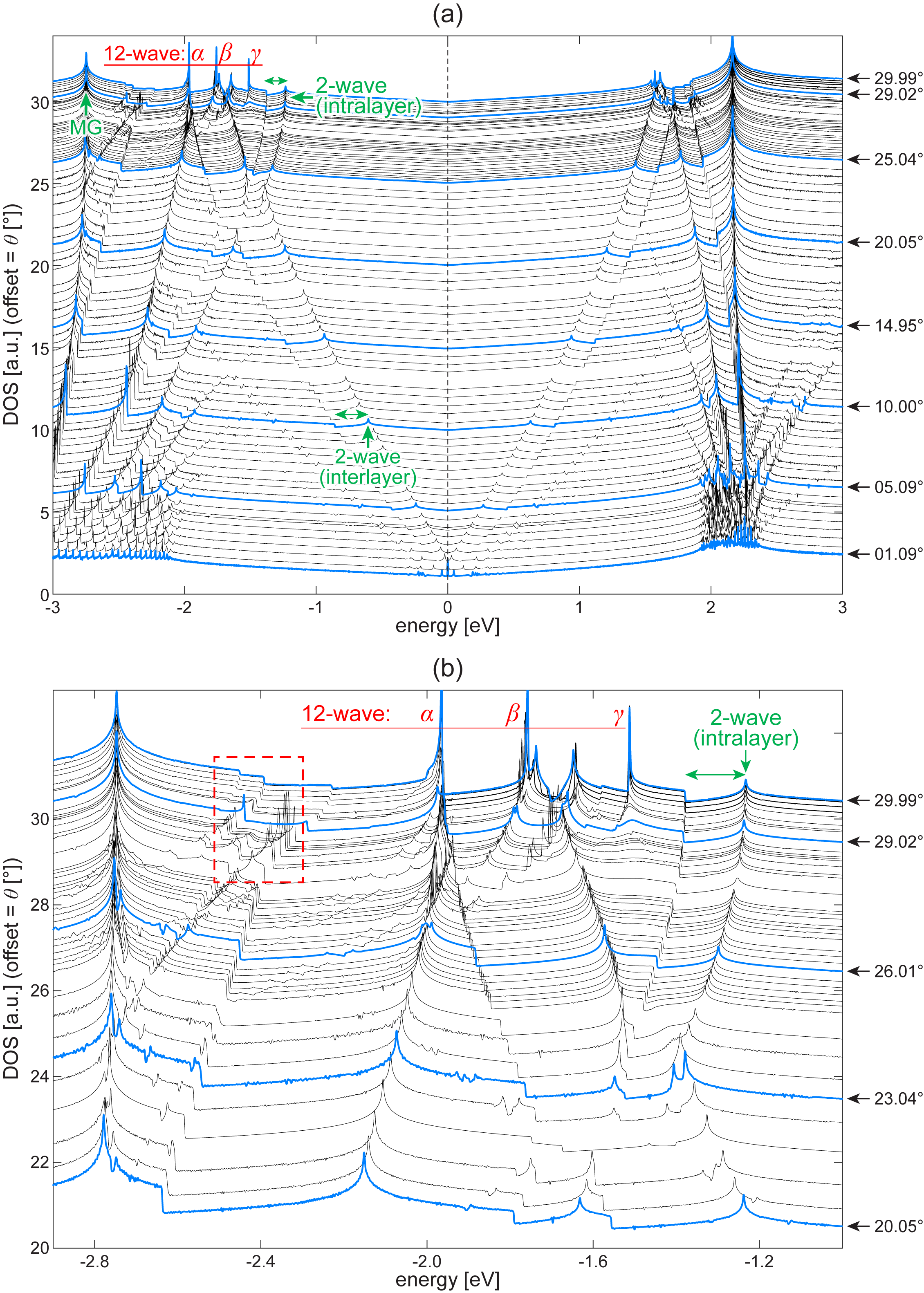 Figure 4
Figure 4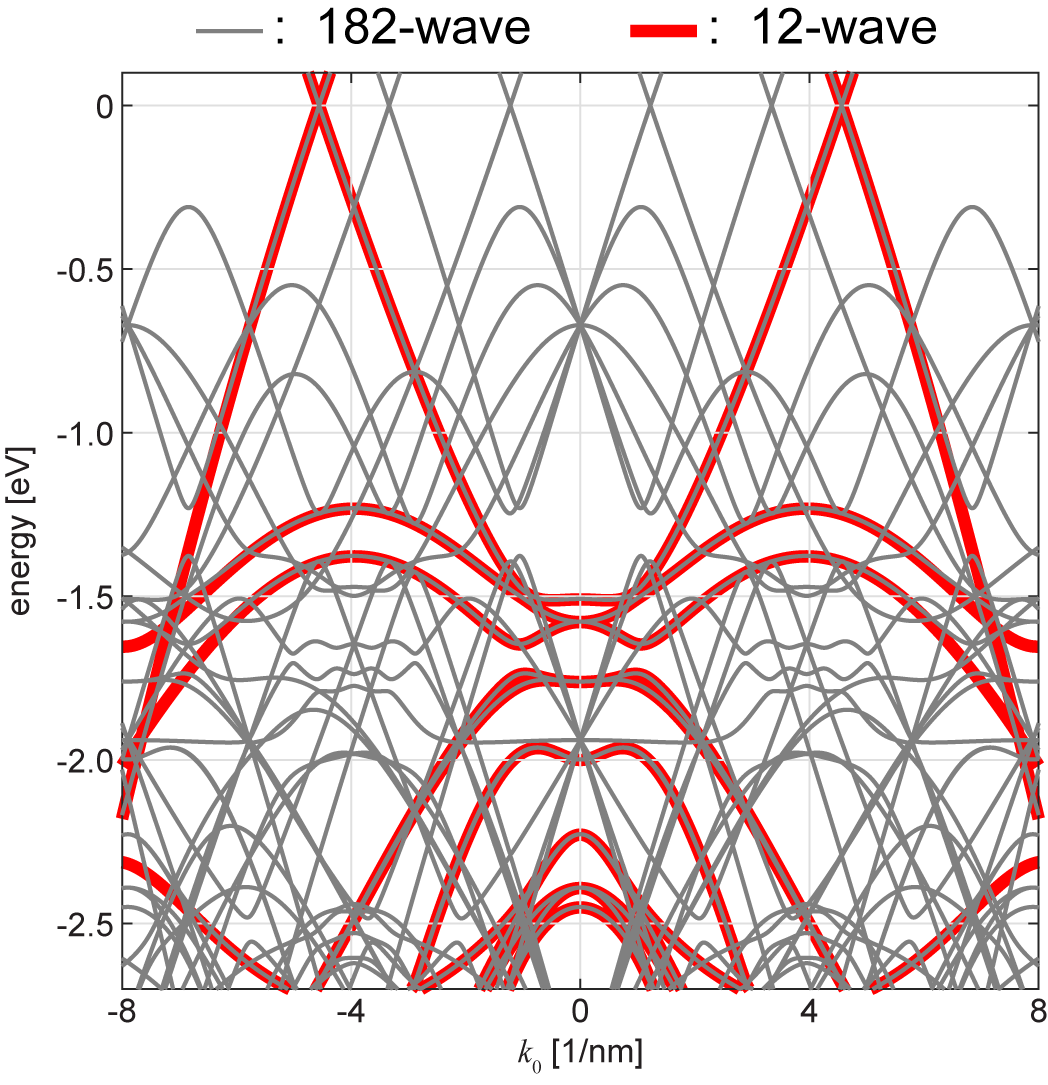 Figure 5
Figure 5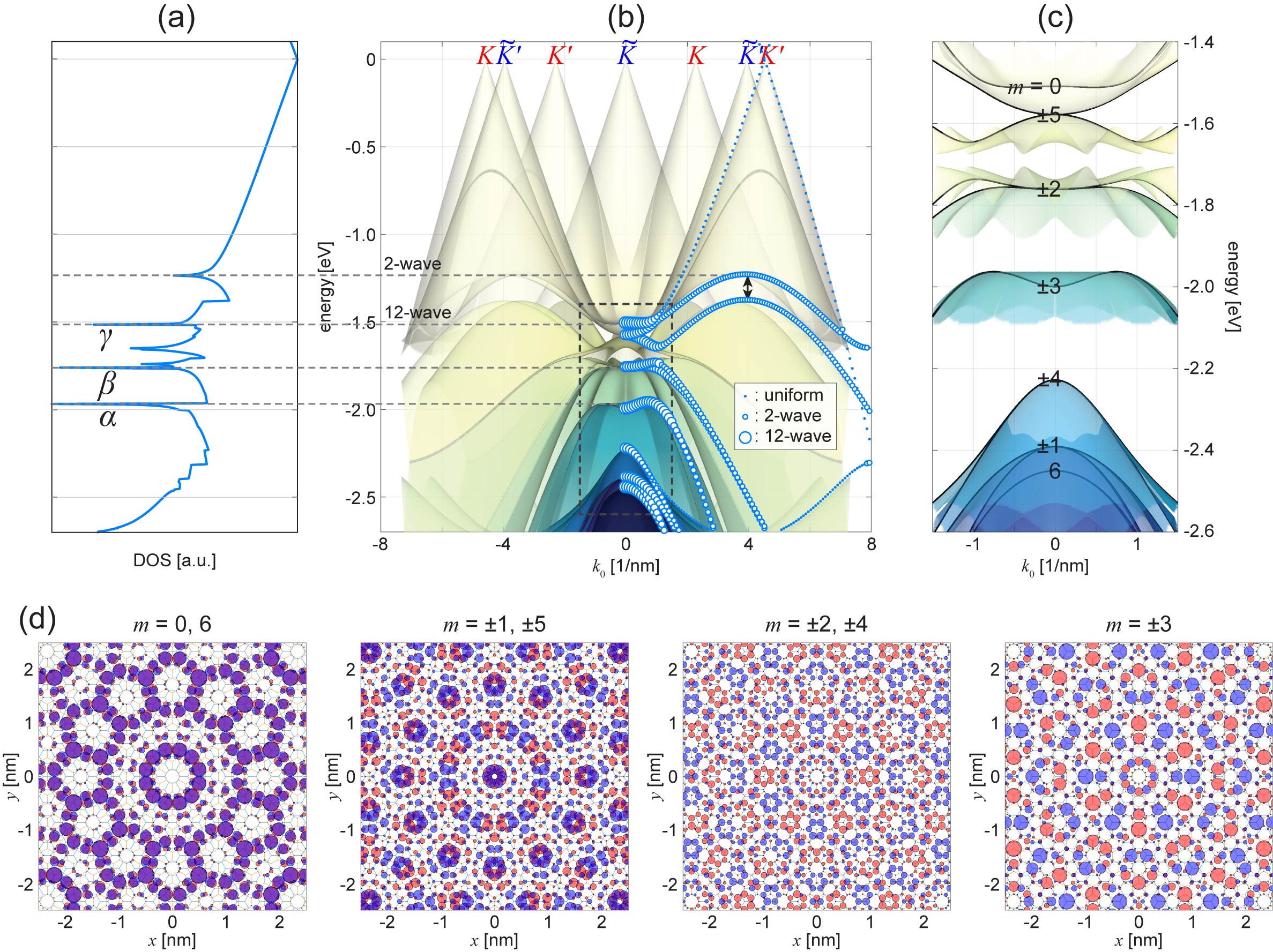 Figure 6
Figure 6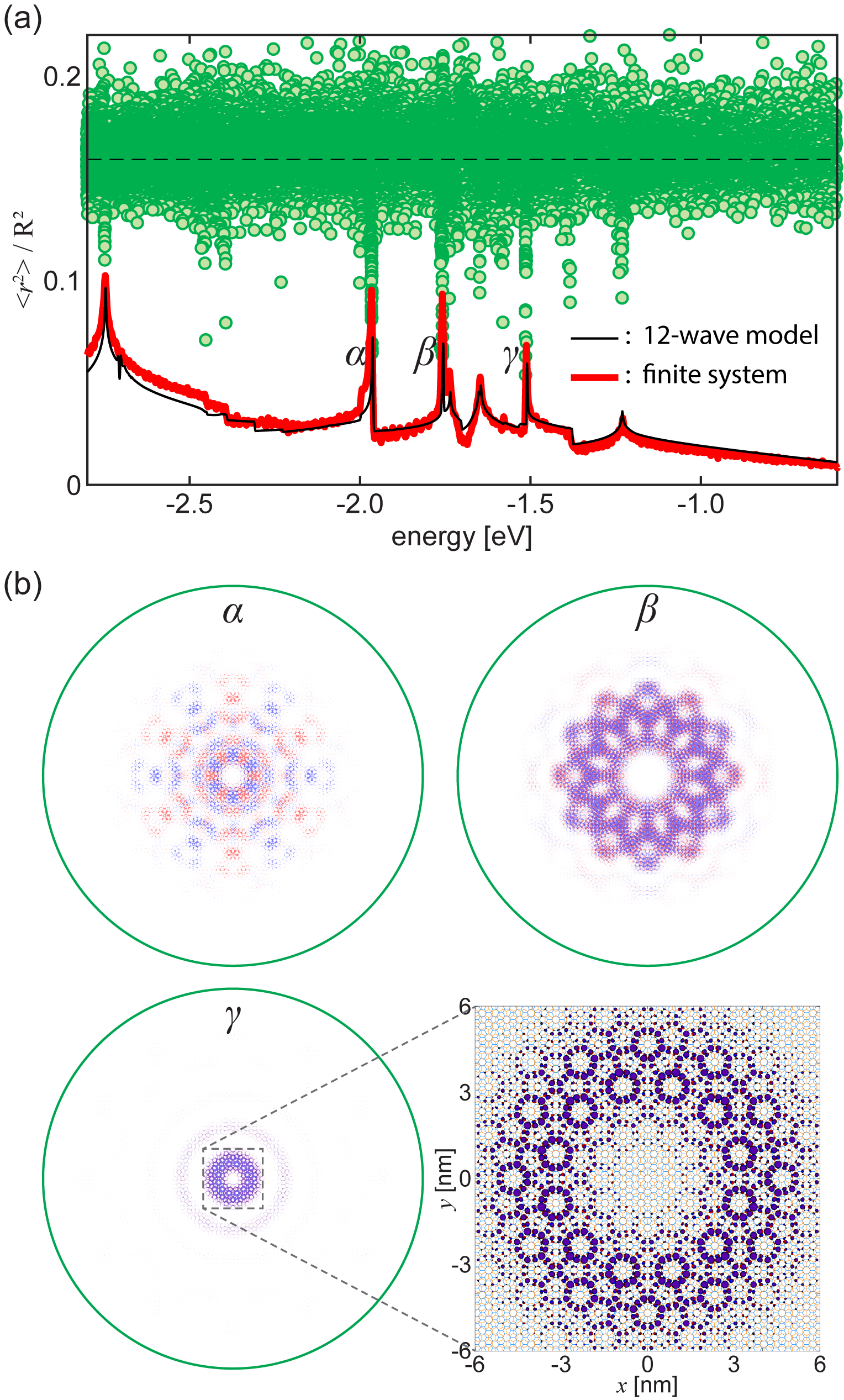 Figure 7
Figure 7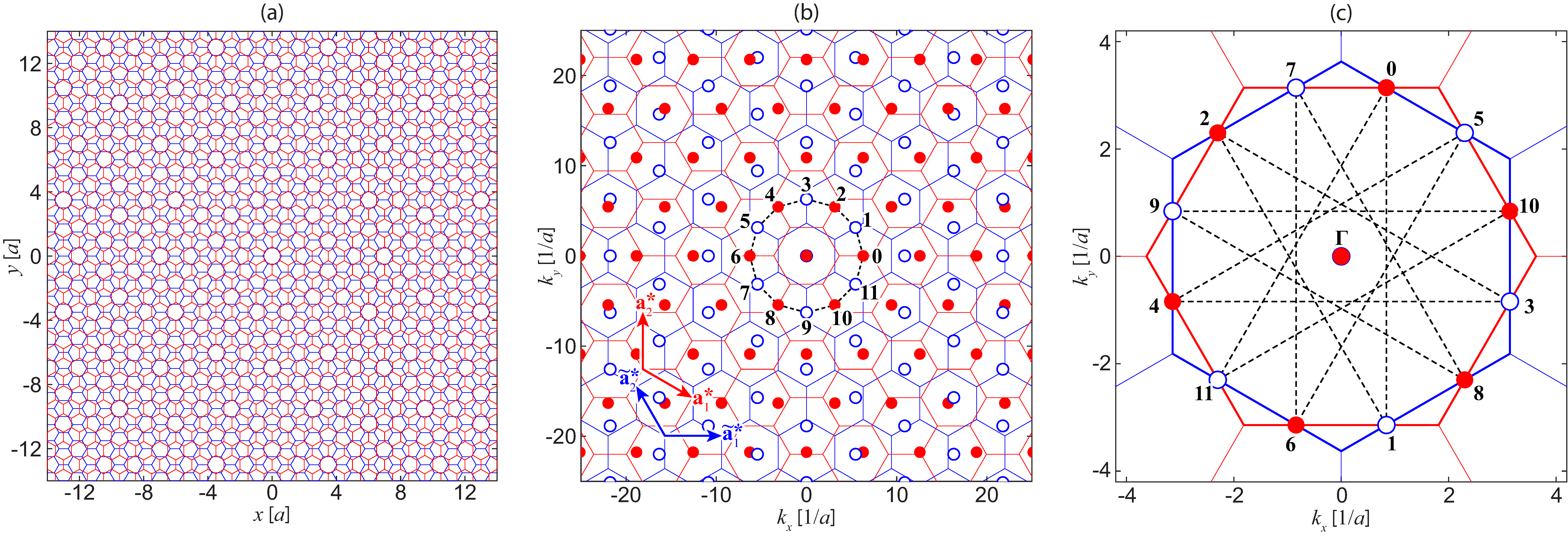 Figure 8
Figure 8Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
††thanks: These authors contributed to the manuscript extensively; Corresponding author: [email protected]††thanks: These authors contributed to the manuscript extensively; Corresponding author: [email protected]
Quasicrystalline electronic states in 30∘ rotated twisted bilayer graphene
Pilkyung Moon
Arts and Sciences, NYU Shanghai, Shanghai, China; NYU-ECNU Institute of Physics at NYU Shanghai, Shanghai, China
Department of Physics, New York University, New York, USA
State Key Laboratory of Precision Spectroscopy, East China Normal University, Shanghai, China
Mikito Koshino
Department of Physics, Osaka University, Toyonaka, Japan
Young-Woo Son
Korea Institute for Advanced Study, Seoul, Korea
Abstract
The recently realized bilayer graphene system with a twist angle of offers a new type of quasicrystal which unites the dodecagonal quasicrystalline nature and graphene’s relativistic properties. Here, we introduce a concise theoretical framework that fully respects both the dodecagonal rotational symmetry and the massless Dirac nature, to describe the electronic states of the system. We find that the electronic spectrum consists of resonant states labeled by 12-fold quantized angular momentum, together with the extended relativistic states. The resulting quasi-band structure is composed of the nearly flat bands with spiky peaks in the density of states, where the wave functions exhibit characteristic patterns which fit to the fractal inflations of the quasicrystal tiling. We also demonstrate that the 12-fold resonant states appear as spatially-localized states in a finite-size geometry, which is another hallmark of quasicrystal. The theoretical method introduced here is applicable to a broad class of “extrinsic quasicrystals” composed of a pair of two-dimensional crystals overlaid on top of the other with incommensurate configurations.
I Introduction
When two graphene layers are overlapped on top of the other, the interlayer twist angle is an important physical quantity to determine the electronic structures. This twisted bilayer graphene (TBG) is essentially a quasi-periodic system, as the two lattice periods of individual graphene layers are generally irrational to each other. When is relatively small (less than about 10∘), however, the low-energy physics is governed by the long-range moiré interference pattern, and then the electronic properties are captured by the moiré effective theory that does not need an exact lattice matching. In brief, the effective theory approximately treats TBG as a translationally-symmetric system ruled by the moiré period. The exotic phenomena in the low-angle regime Mele (2010); Fu et al. (2018), such as the flat band formation Lopes Dos Santos et al. (2007); Trambly de Laissardière et al. (2010); Shallcross et al. (2010); Suárez Morell et al. (2010); Bistritzer and MacDonald (2011); Moon and Koshino (2012); Trambly de Laissardière et al. (2012) and the Hofstadter butterfly under magnetic field Moon and Koshino (2012); Dean et al. (2013); Hunt et al. (2013); Ponomarenko et al. (2013); Moon and Koshino (2014), can be understood in terms of the moiré effective theory.
In TBG of large , on the other hand, the moiré period competes with the atomic length scale and the quasi-periodic nature emerges Koren and Duerig (2016). When , in particular, the overlaid two hexagonal lattices is mapped onto a 12-fold rotationally symmetric quasicrystalline lattice without any translational symmetry [Fig. 1(a)], as first shown by Stampfli Stampfli (1986). Recently, the TBG with a precise rotation angle of was experimentally realized and its spectrum measured in epitaxially grown samples on top of SiC surface Ahn et al. (2018). In addition, similar TBGs have been realized on top of Ni surface Takesaki et al. (2016); Yao et al. (2018) and also by a transfer method Chen et al. (2016). Moreover, another -rotated stack of atomic layers have also been realized in graphene on top of BN layer Wang et al. (2016) as well as MoSe2 bilayer system Choi et al. (2017). In such the quasicrystalline TBG (QC-TBG), the moiré effective approach sketched above breaks down because its main assumption that the moiré pattern governs the system is no longer valid.
In the literature, several theoretical approaches have been applied to understand the electronic structures of conventional quasi-periodic systems Roche et al. (1997), such as one-dimensional Fibonacci lattices Niu and Nori (1986); Kohmoto et al. (1987), two-dimensional non-periodic tiling including Penrose lattice Niizeki and Akamuatsu (1990); Gambaudo and Vignolo (2014), metal nanoparticles Dong et al. (2009), photonic quasicrystals Mnaymneh and Gauthier (2007) and three-dimensional alloys including Al-Mn, Al-Ni-Co, and Al-Cu-Co Smith and Ashcroft (1987); Fujiwara and Yokokawa (1991); Hafner and Krajčí (1992); Trambly de Laissardière and Fujiwara (1994); Roche and Fujiwara (1998); Rogalev et al. (2015). These systems can be viewed as intrinsic quasicrystals where the atomic sites are intrinsically arranged in the quasi-periodic order. In contrast, the QC-TBG is regarded as an extrinsic quasicrystal, in that it is composed of a pair of perfect crystals having independent periodicities, and the quasi-periodic nature appears only in the perturbational coupling between the two subsystems. Thus, the QC-TBG unites the quasicrystalline order and the relativistic nature of the massless Dirac particles of graphene, yet it is not obvious whether and in what form the essential features of quasicrystals emerge in the electronic properties. Since such a hybrid situation is out of the scope of the previous theories of intrinsic quasicrystals, we need an alternative theoretical framework to properly describe the quasicrystalline physics of QC-TBG.
In this paper, we develop a concise model Hamiltonian that fully respects both the dodecagonal rotational symmetry and the massless Dirac nature, to describe the quasicrystalline electronic states in the QC-TBG. We find that the electronic spectrum of QC-TBG is characterized by the 12-fold resonant states of relativistic Dirac fermions, and they can be well captured by a ring Hamiltonian composed of 12 Dirac cones. The resulting quasi-band structure comprises a series of the nearly flat bands corresponding to the resonant states, each of which is labeled by a 12-fold quantized angular momentum. The spatial pattern of wave functions exhibit the fractal inflations of the Stampfli tiling, which is a direct manifestation of the quasicrystalline nature Niizeki (1989). Since we can tune the twist angles in the model, the transition of electronic states from the approximants Goldman and Kelton (1993) of QC-TBG to a true dodecagonal rotational symmetry can be continuously described within a 12-fold ring model, and the emergence of quasicrystalline states and the validity of the approximant method are critically attested. We also show that the 12-fold resonant states appear as spatially-localized states in a finite-size geometry, which is another hallmark of the quasicrystalline nature Niu and Nori (1986); Kohmoto et al. (1987); Deguchi et al. (2012). The proposed theoretical approach is applicable to a broad class of extrinsic quasicrystals, and its simple structure of the closed Hamiltonian allows rigorous analysis on exotic quantum phenomena of quasicrystals.
The paper is organized as follows. In Sec. II, we present the tight-binding model for QC-TBG, and introduce the dual tight-binding approach in the momentum space. In Sec. III.1, we derive the approximate 12-wave ring Hamiltonian, and using this, we describe the quasi-band structure, the resonant states and the characteristic wave functions to respect the Stampfli tiling. In Sec. III.3, we calculate the electronic states of QC-TBG in an alternative method using the finite-size tight-binding model, and demonstrate the localization nature of the 12-fold resonant states. A brief conclusion is given in Sec. IV.
II Theoretical methods
II.1 Tight-binding Hamiltonian for QC-TBG
We define the atomic structure of QC-TBG by starting from AA-stacked bilayer graphene (i.e. perfectly overlapping honeycomb lattices) and rotating the layer 2 around the center of hexagon by [Fig. 1(a)]. We set coordinates parallel to the graphene layers and axis perpendicular to the plane. The system belongs to the symmetry group , and it is invariant under an improper rotation , where is the rotation by an angle around axis, and is the mirror reflection with respect to plane. The primitive lattice vectors of layer 1 are taken as and with the lattice constant , and those of the layer 2 as . Accordingly, the reciprocal lattice vectors of layer 1 are given by and , and layer 2 by . The atomic positions are given by
[TABLE]
where and are integers, () denotes the sublattice site of layer 1(2), and \mbox{\boldmath\tau}_{X} and \mbox{\boldmath\tau}_{\tilde{X}} are the sublattice positions in the unit cell, defined by \mbox{\boldmath\tau}_{A}=-\mbox{\boldmath\tau}_{1}, \mbox{\boldmath\tau}_{B}=\mbox{\boldmath\tau}_{1}, \mbox{\boldmath\tau}_{\tilde{A}}=-R(\pi/6)\mbox{\boldmath\tau}_{1}+d{\bf e}_{z}, \mbox{\boldmath\tau}_{\tilde{B}}=R(\pi/6)\mbox{\boldmath\tau}_{1}+d{\bf e}_{z} with \mbox{\boldmath\tau}_{1}=(0,a/\sqrt{3}). Here is the interlayer spacing between graphene layers and is the unit vector normal to the layer.
We model graphene by the tight-binding model of carbon orbitals. The Hamiltonian is spanned by the Bloch bases of orbitals at difference sublattices,
[TABLE]
where is the atomic orbital at the site , and are the two-dimensional Bloch wave vectors and is the number of graphene’s unit cells in the total system area . We assume that the transfer integral between any two orbitals is expressed as Slater and Koster (1954)
[TABLE]
where is the relative vector between two atoms, , , and Trambly de Laissardière et al. (2010); Moon and Koshino (2013a).
The total tight-binding Hamiltonian is expressed as where and are the Hamiltonian for the intrinsic monolayer graphenes of layer 1 and 2, respectively, and is for the interlayer coupling. The intralayer matrix elements of layer 1 are given by
[TABLE]
where and \mbox{\boldmath\tau}_{X^{\prime}X}=\mbox{\boldmath\tau}_{X^{\prime}}-\mbox{\boldmath\tau}_{X}. Similarly, the matrix for is given by replacing with .
The interlayer matrix element between layer 1 and 2 is written as Mele (2010); Bistritzer and MacDonald (2011); Koshino (2015)
[TABLE]
where and () run over all the reciprocal points of layer 1 and 2, respectively. We also defined
[TABLE]
where z_{\tilde{X}X}=(\mbox{\boldmath\tau}_{\tilde{X}}-\mbox{\boldmath\tau}_{X})\cdot{\bf e}_{z}.
II.2 Dual tight-binding lattice in momentum space
Equation (5) shows that the interlayer interaction occurs between the states satisfying the generalized Umklapp scattering condition . When we start from the layer 1’s Bloch states at , for example, the interlayer Hamiltonian couples this state with layer 2’s Bloch states at . They are further coupled back to layer 1’s states at , and so forth. As a result, the space of the wave functions associated with is spanned by and for and . However, we actually need only a subset of these sets, since the BZ of each layer is translationally invariant with respect to the reciprocal lattice vectors of its own (i.e., and stand for the same Bloch wavenumber of layer 1). Thus, without loss of generality, we can choose the subspace spanned by the QC-TBG Hamiltonian as and . Here note that the -points in each layer is regularly spaced with the reciprocal vectors of the other layer.
According to Eq. (5), the interaction strength between and is given by where . Since decays in large , the relevant contribution occurs only when is relatively small. The interaction strength can be visualized by the diagram Fig. 1(b), where all the layer 2’s wave points are inverted to , and overlapped with the layer 1’s wave points . In the map, the quantity is the geometrical distance by given two points, so that the interaction takes place only between the points located in close distance. If the -points are viewed as ‘sites’, the whole system can be recognized as a tight-binding lattice in -space, which is the dual counterpart of the original tight-binding Hamiltonian in the real space. It should be noted that, unlike the real-space version, the intralayer Hamiltonians and now can be interpreted as -dependent on-site potential in the -space, which is nothing but the band energy of intrinsic graphene. Recently, the relationship between the real space and the momentum space was also noticed in the localized wave functions in moiré bilayer systems Carr et al. (2018).
In this -space tight-binding model, the hopping between different -space sites (the interlayer interaction ) is smaller by an order of magnitude than the potential landscape (the band energy), so that the eigen functions tend to be localized in the -space lattice, in a similar manner to the Aubry-André model in one dimension Aubry and André (1980). In the practical calculation, therefore, we are allowed to take only a limited number of wave points around inside a certain cut-off circle, and obtain the energy eigenvalues by diagonalizing the Hamiltonian matrix within the finite bases. If we plot the energy levels against , we obtain the quasi-band structures of the system. Here the wavenumber works like the crystal momentum for the periodic system, so it can be called the quasicrystal momentum. The cut-off radius should be greater than the typical localization length in the -space, but need not be too large, since the wave points discarded outside are properly considered by shifting . If we increase , we will see more and more replicas of the identical quasi-energy band with different origins, because shifting actually corresponds to taking a different origin in the -space map of Fig. 1(b). The resonant band structure near barely changes in this process because its wave function is very well localized to the 12-membered ring in the -space. The replica bands are just duplication of the same states so they should be appropriately removed in calculating the physical quantities such as the density of states. The validity of the momentum cut-off is discussed in detail in Appendix A.
III Results and discussion
III.1 12-fold symmetric resonant states
At , we see that the twelve symmetric points form a circular chain in the dual-tight-binding lattice of which radius is , indicated by the dashed ring in Fig. 1(b). Noting that the layer 2’s wave points are inverted, these points are associated with layer 1’s Bloch wavenumbers for even ’s and layer 2’s for odd ’s. Figure 1(c) shows the original positions of (layer 1) and (layer 2) associated with , in the first Brillouin zone. Due to the symmetry, the intrinsic graphene’s Bloch states at the twelve points are all degenerate in energy, and therefore the interlayer coupling hybridizes them to make resonant states. Here the coupling is only relevant between the neighboring sites of the ring, and it is given by .
In the vicinity of , the Hamiltonian of the ring can be expressed by a matrix,
[TABLE]
where , , and we neglect the dependence of the interlayer matrix element . The diagonal block represent monolayer’s Hamiltonian at for even (layer 1) and for odd (layer 2). In each block the sublattices are arranged in the order of or for in modulo of 4, and or for . By doing this, the first base of a block is always mapped to the first base of other block under the operation of . Note that the arrangement of , , etc. in the submatrix is fixed irrespective of , and the dependence on solely comes from in the argument of . Consequently, the total Hamiltonian is obviously symmetric under rotation by a single span of the ring (i.e., moving to ), which actually corresponds to the operation (210∘ rotation and swapping layer 1 and 2) in the original system.
Figures 2 show (a) the density of states (DOS), (b) the band structures as a function of in the negative energy region, and (c) its closer view near . The twelve Dirac cones are arranged on a circle with a radius and they are strongly mixed near . As a result, the originally degenerate twelve states of graphene (in each of the electron side and the hole side of the Dirac cone) split into different energies, and exhibit the characteristic dispersion including flat band-bottoms and the Mexican-hat edges. This leads to a series of spiky peaks and dips (pseudogaps) in DOS. At , the Hamiltonian can be analytically diagonalized to obtain a set of energies (neglecting the constant energy)
[TABLE]
where 1.84 eV, corresponds to the conduction band and valence band, respectively, and with is the wavenumber along the chain. The eigenvalue of is given by . Here the states with form twofold doublets, and belong to two-dimensional irreducible representation of point group. The and are non-degenerate, and belong to and , respectively, for the conduction (valence) band. If we disregard the -position difference, the index is regarded as quantized angular momentum in 12-fold rotational symmetry, and this is an essential characteristics of quasicrystal TBG.
We have similar resonant states also in the conduction band, while the energy scale of the band structures is much smaller than in the valence band. Equation (9) clearly explains such asymmetry; the dispersion of in is nearly three times wider than that of , considering that . Intuitively, the wave function of the conduction band of intrinsic graphene has the opposite phases between the sublattice and , and then the interlayer coupling between incommensurate layers is tend to be suppressed by the phase cancellation.
III.2 Wave functions showing the quasicrystal tiling
The 12-wave resonant coupling also gives rise to a characteristic pattern in the wave function. Figure 2(d) shows the wave functions at where the hybridization is the most prominent, where we can see that the wave amplitude distribute selectively on a limited number of sites in a 12-fold rotationally symmetric pattern. The extent of the hybridization of different wave modes is characterized by the inverse participation ratio (IPR) on the dominant layer,
[TABLE]
Here is the amplitude at the site of the eigenstates , and represents the sum over the sites on the dominant layer, which is defined as the layer having greater wave amplitude than the other. We have for a pure single layer state, and for a hybrid state of two plain wave modes. In Fig. 2(b), the blue dots represent IPR at several sample points in the band structures along direction, where the dot area is proportional to . We see that the IPR becomes large exclusively around , where the 12 wave components are strongly hybridized. remains almost 1 near the Dirac cones where the hybridization is almost negligible. We also have a region of along the arch-shaped gap below the Dirac cone, which is indicated by “2-wave” in Fig. 3(b). These “2-wave” states arise from the hybridization of the and of the same layer assisted by the second-order process of the interlayer coupling .
We also show a large scale plot of states in Fig. 3(a). We see that the wave pattern perfectly follows the Stampfli tiling, where the red (left-half) and black (right-half) lines represent the third and fourth generations of the fractal inflation, respectively Stampfli (1986). Such the long-range structure of the quasicrystalline wave function is actually quite sensitive to a slight change of the twist angle. Figure 3(b) represents the wave pattern of the corresponding state in TBG with , calculated by the same 12-wave method. The TBG of is a quasicrystal approximant, which is not quasi-periodic but has a translational symmetry with period of . We can see that the local wave pattern is quite similar to that of , while the long-range quasi-periodic nature is completely lost and round to a periodic pattern. Here we confirmed that the quasi-band structures, DOS and IPR look almost the same as , but the tiny change of the wave bases and the coupling matrix elements in the 12-ring Hamiltonian encodes the periodic / quasi-periodic transition.
The energy spectrum of the QC-TBG approximant can also be calculated by the original real-space tight-binding model since it has a finite superlattice unit cell. We can show that the DOS and the wave function of calculated by the original tight-binding model are virtually the same as the result of the 12-ring effective Hamiltonian, and this justifies the validity the effective approach. In the appendix B, we present the extensive study on the electronic structures of the quasi approximants in all the angle region from to .
III.3 Localization in finite-sized QC-TBG
The emergence of quasicrystalline states in QC-TBG can also be confirmed by a finite-sized tight-binding lattice, while the computation is enormous. Here we consider a tight-binding lattice composed of two large disks of graphene with radius stacked at exactly , and calculate its electronic structures by diagonalizing the huge Hamiltonian matrix with the total number of atoms 371,532. As shown in Fig. 4(a), the DOS of the finite flakes (thick red line), which is obtained by broadening its discrete spectrum, is consistent with the DOS of the 12-wave effective model (thin black line) calculated by the effective Hamiltonian with a few wave bases [Fig. 2(b)]. In Fig. 4(b), we also present the wave functions at three energies, , and , which correspond to the band edges of the quasi-band structures in the effective Hamiltonian (Fig. 2).
The magnified plot of is presented in the inset of Fig. 4(b) showing the characteristic pattern of 12-wave approximation. Interestingly, however, it is overlapped with an envelope function decaying in the radial direction. Such the localized feature is never seen in single layer graphene and it is the characteristics of the resonant states of QC-TBG. As a measure of the concentration to the center, we calculate the second momentum for each eigenstate and plot it as green circles in Fig. 4(a). For a uniform state (i.e., the wave amplitude is constant throughout the system), approaches , which is indicated by the dashed line. We can actually see that lies around this line for most of the states, while it becomes exceptionally small at the energies of the quasi-band edges argued in the previous section. In terms of the quasi-band structures, these localized states actually correspond to the integral of the quasi-band states over the nearly flat region, and the length scale of the envelope function is related to the size of the flat area in the momentum space. For the state at ( state), for instance, the radius of the flat area is roughly given by , and the corresponding real-space scale matches the characteristic decaying and oscillating scale of the envelope function.
IV Conclusions
We revealed that the quasicrystalline nature emerges in the electronic properties of QC-TBG, or the twisted bilayer graphene stacked at 30∘. We developed a concise model Hamiltonian for this unique system, and demonstrated that the electronic structure is well described by the quasi-band picture despite of the lack of periodicity. The quasi-band states of the QC-TBG are characterized by the 12-fold resonant states of relativistic Dirac fermions, where the wave functions exhibit the spatial pattern fully respecting the dodecagonal quasicrystal tiling. Such a non-uniform distribution of electron may be observed by microscopy imaging techniques. The emergence of quasicrystalline states was attested by comparing the QC-TBG and a periodic approximant near , and it was demonstrated that even a slight deviation from the QC configuration destroys the long-range quasicrystalline nature. Finally, we studied the electronic states of QC-TBG using the finite-size tight-binding model, where the 12-fold resonant states appear as spatially-localized states in a finite-size geometry.
While we considered the QC-TBG as a model example in this paper, the theoretical method based on the -space tight-binding approach introduced here is applicable to any kind of extrinsic quasicrystals composed two-dimensional materials overlaid in incommensurate configurations, including heterostructures of two-dimensional materials having difference lattice symmetries (e.g., rectangle and hexagon).
Extrinsic quasicrystals also provide a unique opportunity to tune the quasicrystal bands by controlling the interlayer interaction strength . As is an exponential function of the interlayer spacing , Koshino et al. (2015) we can either increase by applying pressure, or decrease it through intercalation of ions or addition of barrier atomic layers Chittari et al. (2018). When becomes comparable to the width of the energy bands, we expect a transition from the weakly coupled regime to the strongly coupled regime where the quasicrystalline nature is even more pronounced. The detailed studies on exotic electronic natures in a broad class of extrinsic quasicrystals, such as the electronic transport, optical properties, the quantum Hall effect, and also the effects of modulation to these phenomena, are left for future research.
Acknowledgments
We thank L. A. Wray and A. Kent for fruitful discussions. P.M. was supported by NYU Shanghai (Start-Up Funds), NYU-ECNU Institute of Physics at NYU Shanghai, New York University Global Seed Grants for Collaborative Research. This research was carried out on the High Performance Computing resources at NYU Shanghai and CAC of KIAS. M.K. was supported by JSPS KAKENHI Grant Numbers JP25107005, JP15K21722, JP17K05496. Y.-W.S was supported by NRF of Korea (Grant No. 2017R1A5A1014862, SRC program: vdWMRC center).
Appendix A Validity of the momentum space cut-off
In this section, we argue about the validity of introducing the momentum space cut-off in calculating the quasi band structure. As we mentioned in Sec. II.2, the wave functions of the QC-TBG are localized in the -space in a similar manner to the Aubry-André model in one-dimension Aubry and André (1980), because the hopping term in the -space is much smaller than the potential landscape (the band energy). Figures 5(b)-(d) show some examples of the -space amplitude map. The panel (b) shows a nearly decoupled state which is dominated by only a single state of monolayer graphene, and (c) is a state originating from the 2-wave mixing, where a pair of monolayer’s states on layer 2 are coupled though the mediation of a middle state on layer 1. The panel (d) is the 12-wave resonant state. Any eigenstates other than those examples are also localized within just a few reciprocal lattice constants in -space. When we increase the number of total wave bases components ( and ) by increasing , each eigenstate hardly changes as long as is greater than the typical localization length. In Fig. 5(a), we show the histogram of the number of dominant wave components in the eigenstates at a particular , calculated in the basis sets within of , and , respectively, where the total number of wave bases are , and , respectively. We actually see that each of eigenstates is composed only a few (mostly less than 10) bases. We note that, in large , we often see a resonance between different localized states which are very distant in -space. This does not much affect the calculation of the physical quantity because the overlap of the different localized wave functions are exponentially small.
We have infinitely many localized states far away from the first Brillouin zone, so one might think that it is necessary to take an infinite to properly include all the states. Note that, however, these localized states can be moved into the vicinity of the first Brillouin zone by shifting with a proper amount, as we show in the following. Thus, instead of using a large requiring a large computational cost, we can obtain the full spectrum of the system by calculating the electronic structures as a function of with a moderate .
Let us consider two states and with
[TABLE]
for a given . Suppose and are outside the cut-off circle, i.e., and , but they strongly interact with each other, i.e.,
[TABLE]
Now, for any such , we can always find () which makes move to the point in the first Brillouin zone, i.e.,
[TABLE]
And suppose , defined as
[TABLE]
Then, by shifting to a new point defined as
[TABLE]
we can see that
[TABLE]
are the member of the subspace spanned from . And by considering that , and from Eqs. 12 and 13, we can show that these two points are within the cut-off circle. Since
[TABLE]
and represent the Bloch states same to and , respectively, interacting with the same interaction strength , since
[TABLE]
Thus, by shifting to , the points discarded outside with are properly considered. And by calculating the electronic structures for every in the first Brillouin zone, we can get every possible interaction pairs in this system.
Figure 6 shows the quasicrystal bands of QC-TBG calculated with 12-wave model ( with removed) and 182-wave model (). We can see that the band structure of 182-wave model fully includes the spectrum of 12-wave model, while also contains many other band lines. Actually, these extra lines are just replicas of the identical quasicrystal bands of 12-wave model with different origins (Sec. II.2). In other words, the physical properties can be well described by calculating the quasi-band structure with a relatively short .
The minimum 12-wave models well reproduces the band structure near the 12-wave resonant states, while there are some small errors in the other energies. In Fig. 7, we see that the 182-wave model almost perfectly overlaps with the DOS of very large finite flakes, while 12-wave model slightly under-/overestimates the density of states far from the resonant-state peaks. We confirmed that further increase of does not change the DOS profile.
Appendix B Quasicrystal approximants
In Sec. III.1, we compared the quasicrystalline TBG stacked at (QC-TBG) and its periodic approximant at . Actually there exist infintely many periodic TBGs in any finite region in , just like rational numbers in the real number axis. As we will see the following, the peak structure in the density of states changes almost continuously in rotating the twist angle , and tracing its evolution is useful to get insights on the connection between QC-TBG and the low-angle moiré TBGs, although the computation requires enormous number of atomic bases ( atoms).
Here we calculated the electronic structures of periodic TBGs with various using a tight-binding model. Figure 8(a) shows the evolution of the DOS in a wide range of energy, and Fig. 8(b) is the magnified plot near the critical states of QC-TBG. The peaks marked with “MG” correspond to the van Hove singularity of monolayer graphene. Similarly, “2-wave-interlayer” represents the singularity originates from the two-wave mixing between the states in different layers Moon and Koshino (2013b), and “2-wave-intralayer” is the mixing between the states in the same layer. Koshino et al. (2015); Yao et al. (2018) We show the width of the band opening (pseudogap) from 2-wave interlayer/intralayer mixing by the green horizontal arrows.
The sharp peaks marked as , , correspond to the singularities coming from the 12-wave mixing of QC-TBG, which were described as the nearly flat bands in the quasi-band picture in Sec. III.1. We can see that the singular peaks rapidly grow as approaches . The DOS of the TBG with , the periodic TBG closest to 30∘ in this calculation, is consistent with that of QC-TBG [Fig. 2(a)]. It should be noted that, however, the wave functions of the approximants do not obey the quasicrystalline long-range structure with 12-fold rotational symmetry, as argued in Fig. 3(b). We also see that the peak-and-dip structure in the valence band is much wider than in the conduction band, and it is consistent with the analytic argument in the 12-wave ring model [Eq. (9)].
The peaks enclosed by the red box in Fig. 8(b) are associated with the resonant states other than , , [i.e., the solutions of Eq. (7) other than , , ]. In the 12-wave model, we can also show that corresponding states have flat dispersion in quasi-band structure at , and exhibit singularities in DOS. As approaches , however, the quasi-band becomes dispersive [Fig. 2(b)] and the DOS singularities disappear.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Mele (2010) E. J. Mele, Phys. Rev. B 81 , 161405 (2010).
- 2Fu et al. (2018) Y. Fu, E. König, J. Wilson, Y.-Z. Chou, and J. Pixley, ar Xiv preprint ar Xiv:1809.04604 (2018).
- 3Lopes Dos Santos et al. (2007) J. M. B. Lopes Dos Santos, N. M. R. Peres, and A. H. Castro Neto, Phys. Rev. Lett. 99 , 256802 (2007).
- 4Trambly de Laissardière et al. (2010) G. Trambly de Laissardière, D. Mayou, and L. Magaud, Nano Lett. 10 , 804 (2010).
- 5Shallcross et al. (2010) S. Shallcross, S. Sharma, E. Kandelaki, and O. Pankratov, Phys. Rev. B 81 , 165105 (2010).
- 6Suárez Morell et al. (2010) E. Suárez Morell, J. D. Correa, P. Vargas, M. Pacheco, and Z. Barticevic, Phys. Rev. B 82 , 121407 (2010).
- 7Bistritzer and Mac Donald (2011) R. Bistritzer and A. H. Mac Donald, PNAS 108 , 12233 (2011).
- 8Moon and Koshino (2012) P. Moon and M. Koshino, Phys. Rev. B 85 , 195458 (2012).