Molecular dynamic simulation of water vapor interaction with various types of pores using hybrid computing structures
V.V. Korenkov, E.G. Nikonov, M. Popovi\v{c}ov\'a

TL;DR
This paper explores the efficiency of molecular dynamics simulations for water vapor interacting with different pore types, emphasizing parallel computing to reduce computational time and improve simulation performance.
Contribution
It evaluates various parallel algorithms for MD simulations of water vapor in pores, highlighting improvements over serial computations and analyzing parameter impacts.
Findings
Parallel calculations significantly reduce simulation time.
Simulation efficiency depends on pore shape and particle number.
Parallel algorithms outperform serial methods in MD simulations.
Abstract
Theoretical and experimental investigations of water vapor interaction with porous materials are very needful for various fields of science and technology. Not only studies of the interaction of water vapor and porous material as a continuous medium, but also the study of the interaction of water vapor with individual pore is very important in these researches. Mathematical modelling occupies an important place in these investigations. In this work, a study of efficiency of various implementations algorithms for MD simulation of water vapor interaction with individual pore is carried out. A great disadvantage of MD is its requirement of a relatively large computational effort and long time in simulations. These problems can be drastically reduced by parallel calculations. In this work we investigate dependence of time required for simulations on different parameters, like number of…
Click any figure to enlarge with its caption.
 Figure 1
Figure 1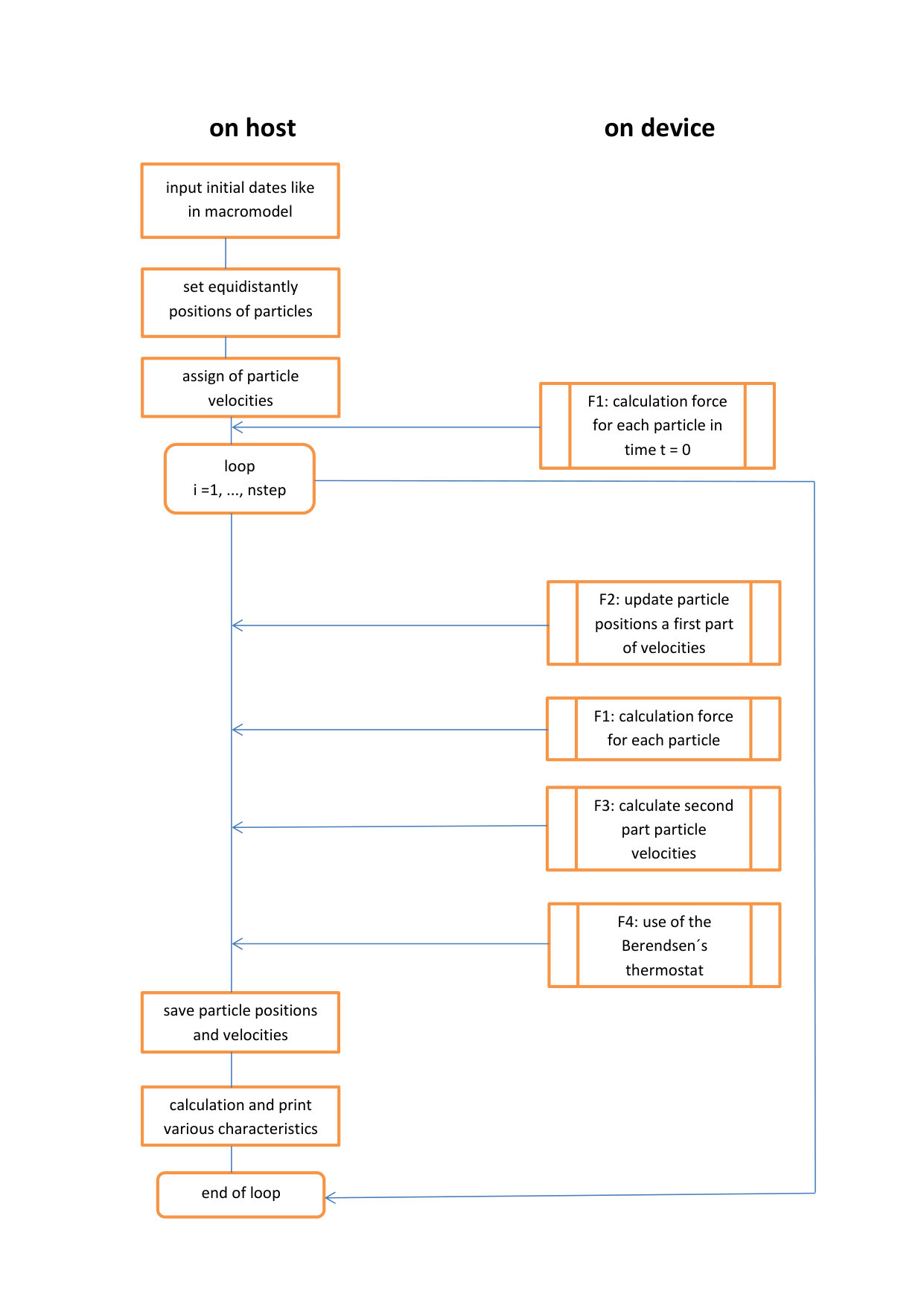 Figure 2
Figure 2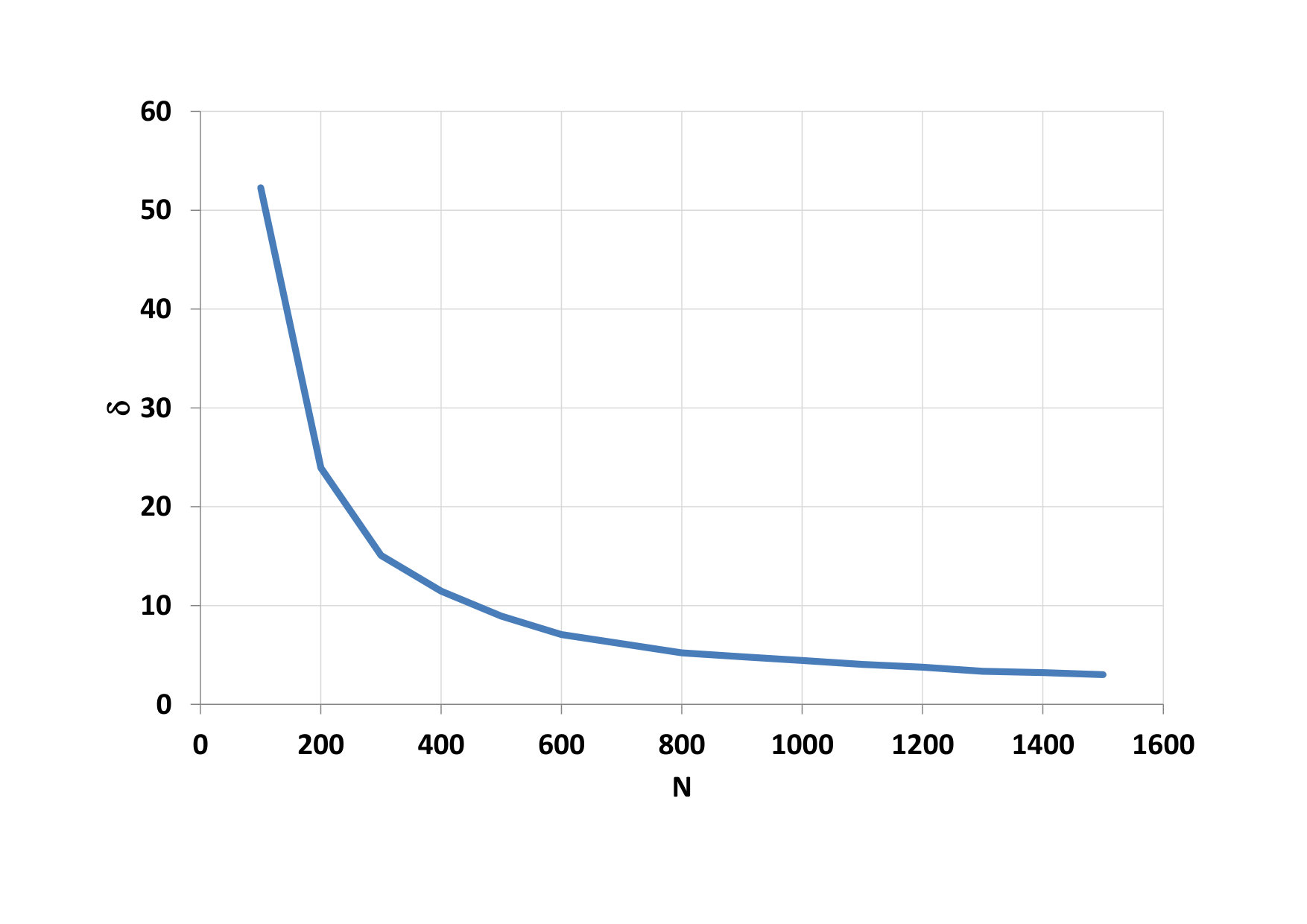 Figure 10
Figure 10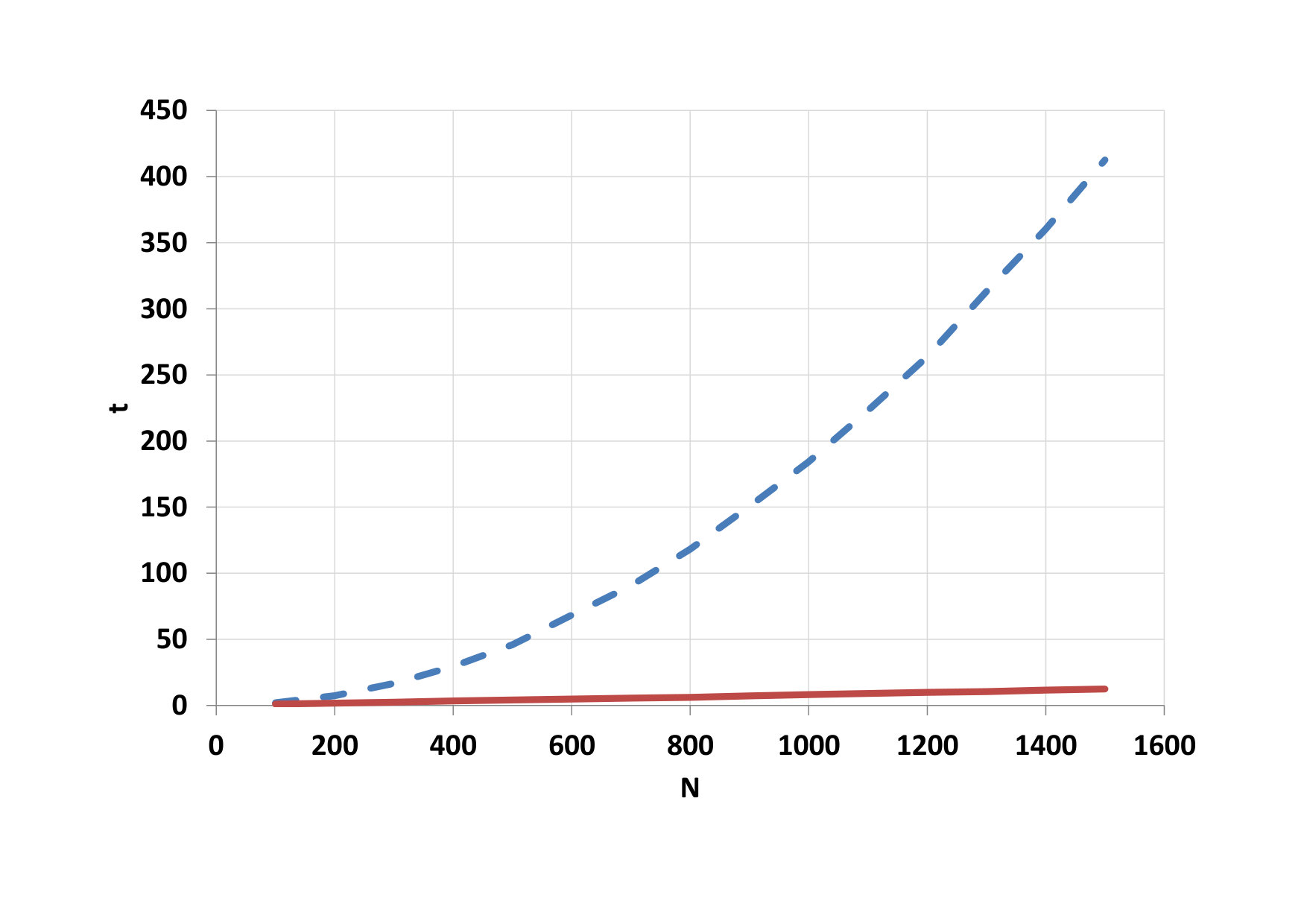 Figure 11
Figure 11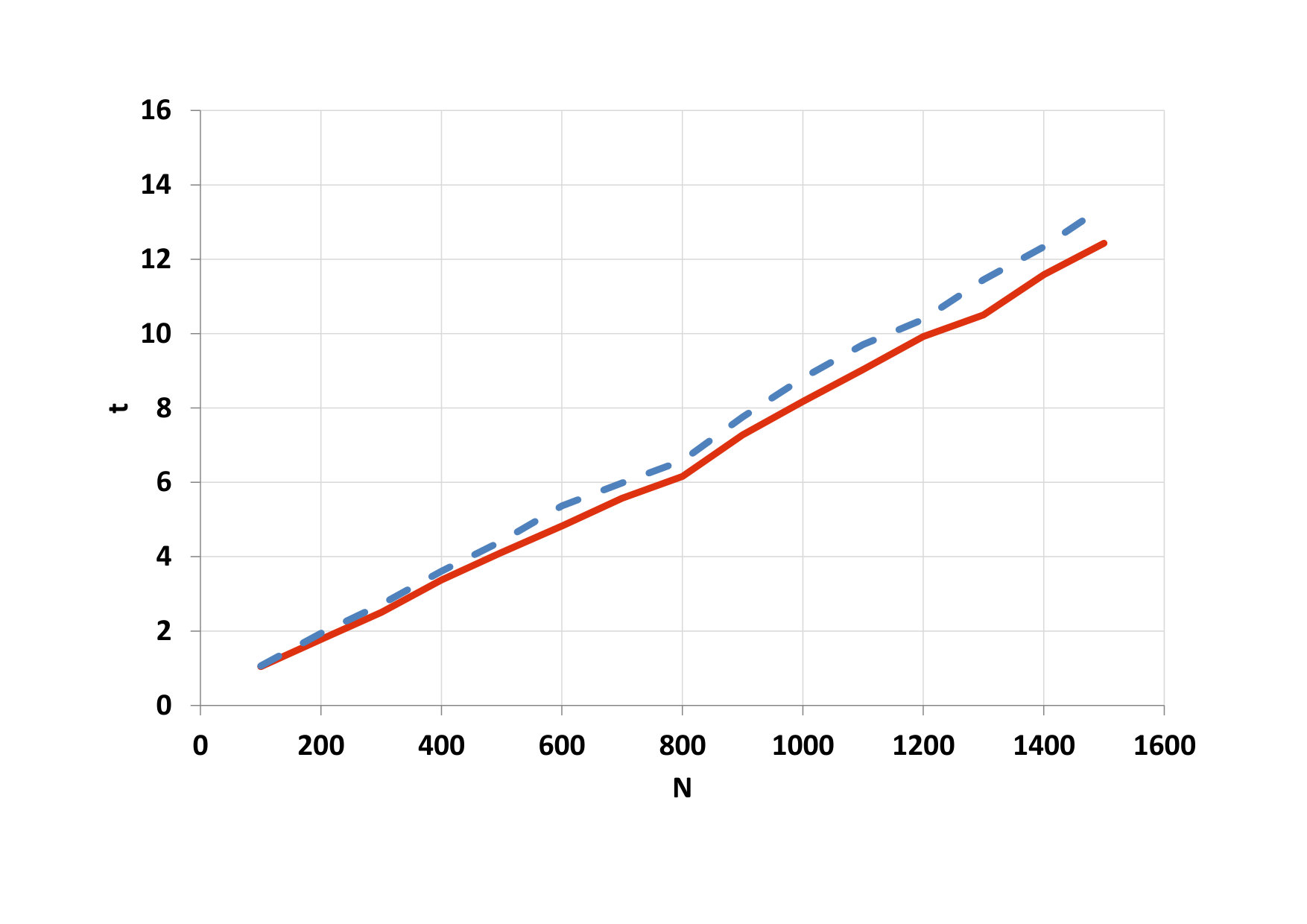 Figure 12
Figure 12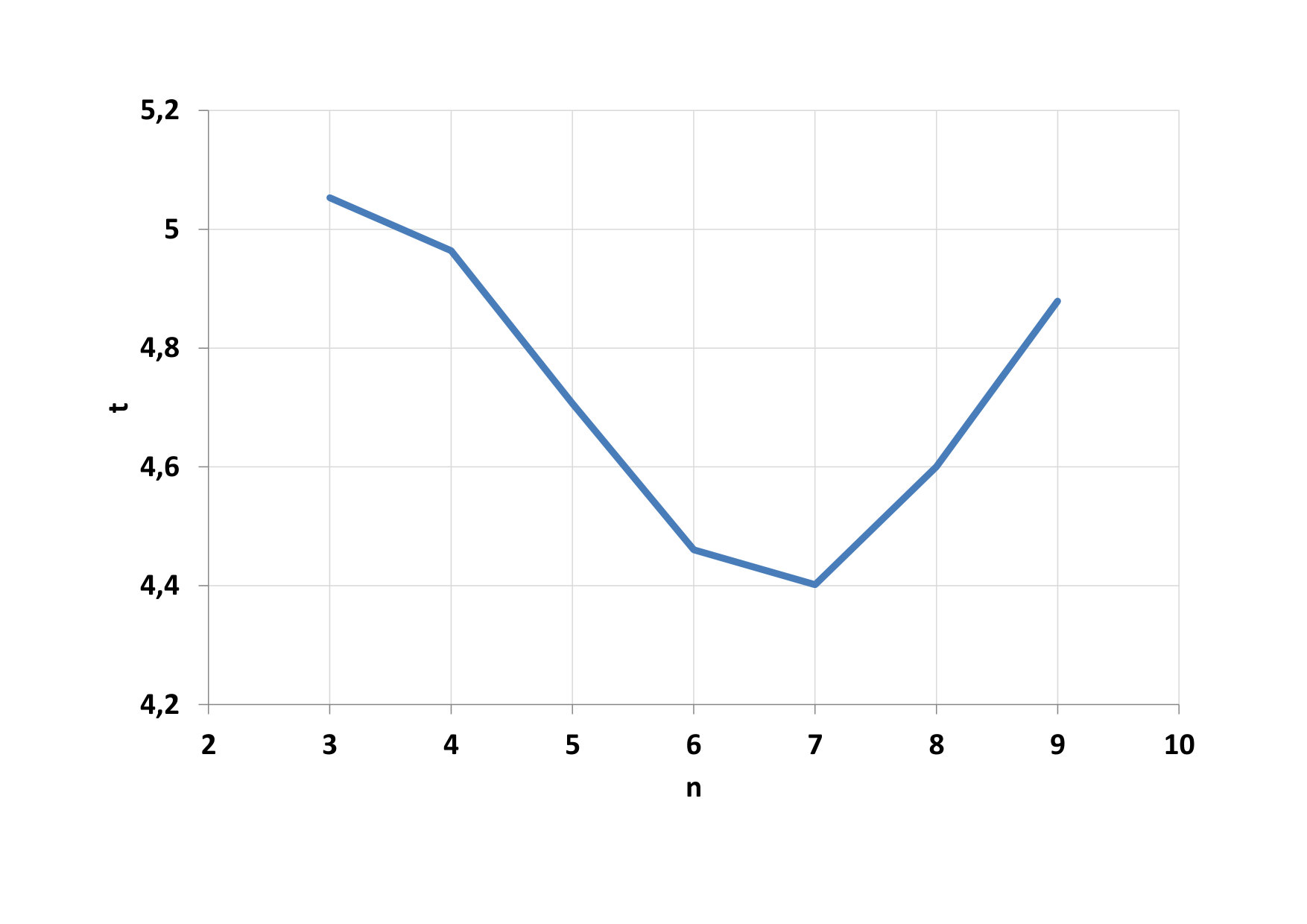 Figure 2
Figure 2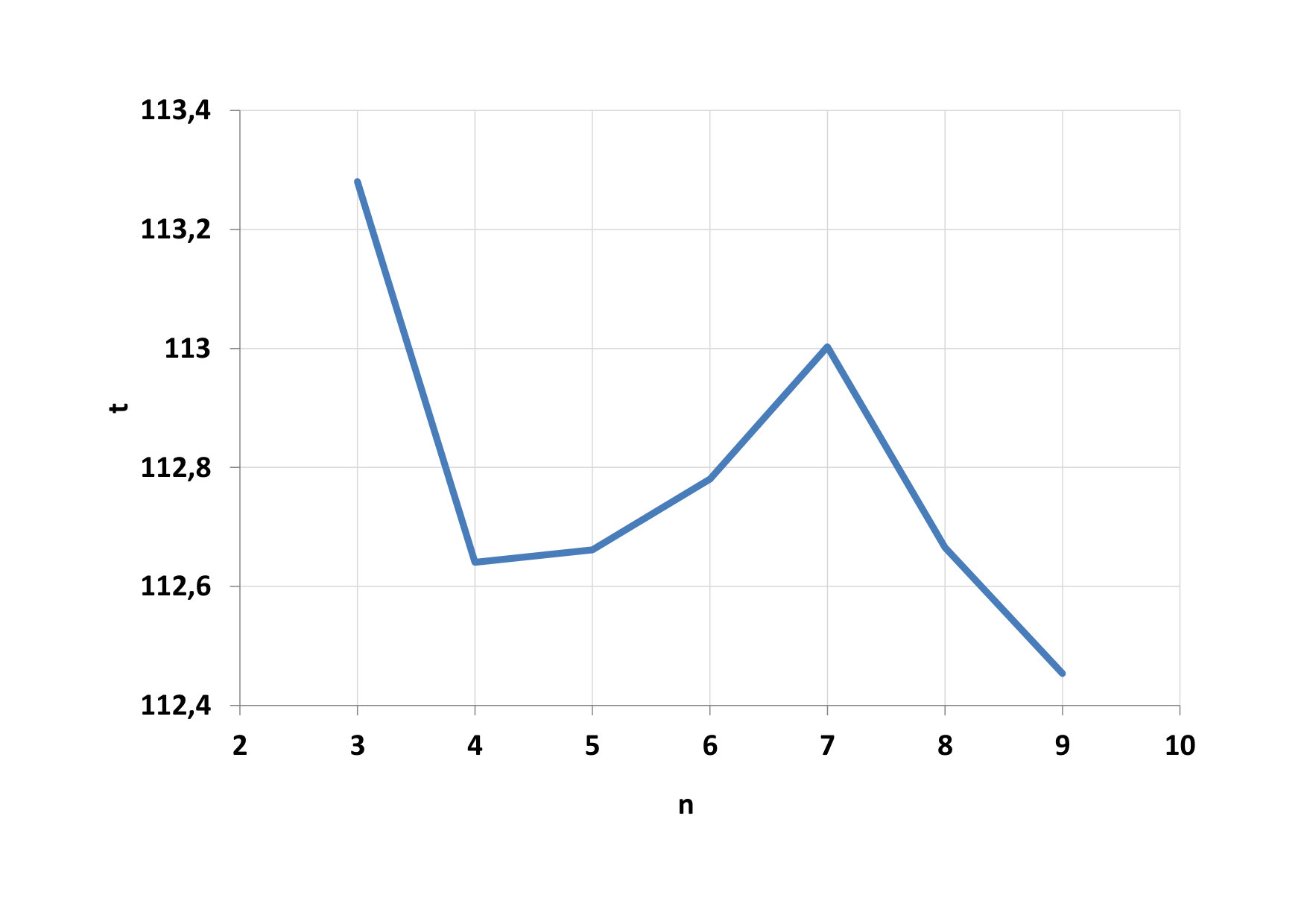 Figure 3
Figure 3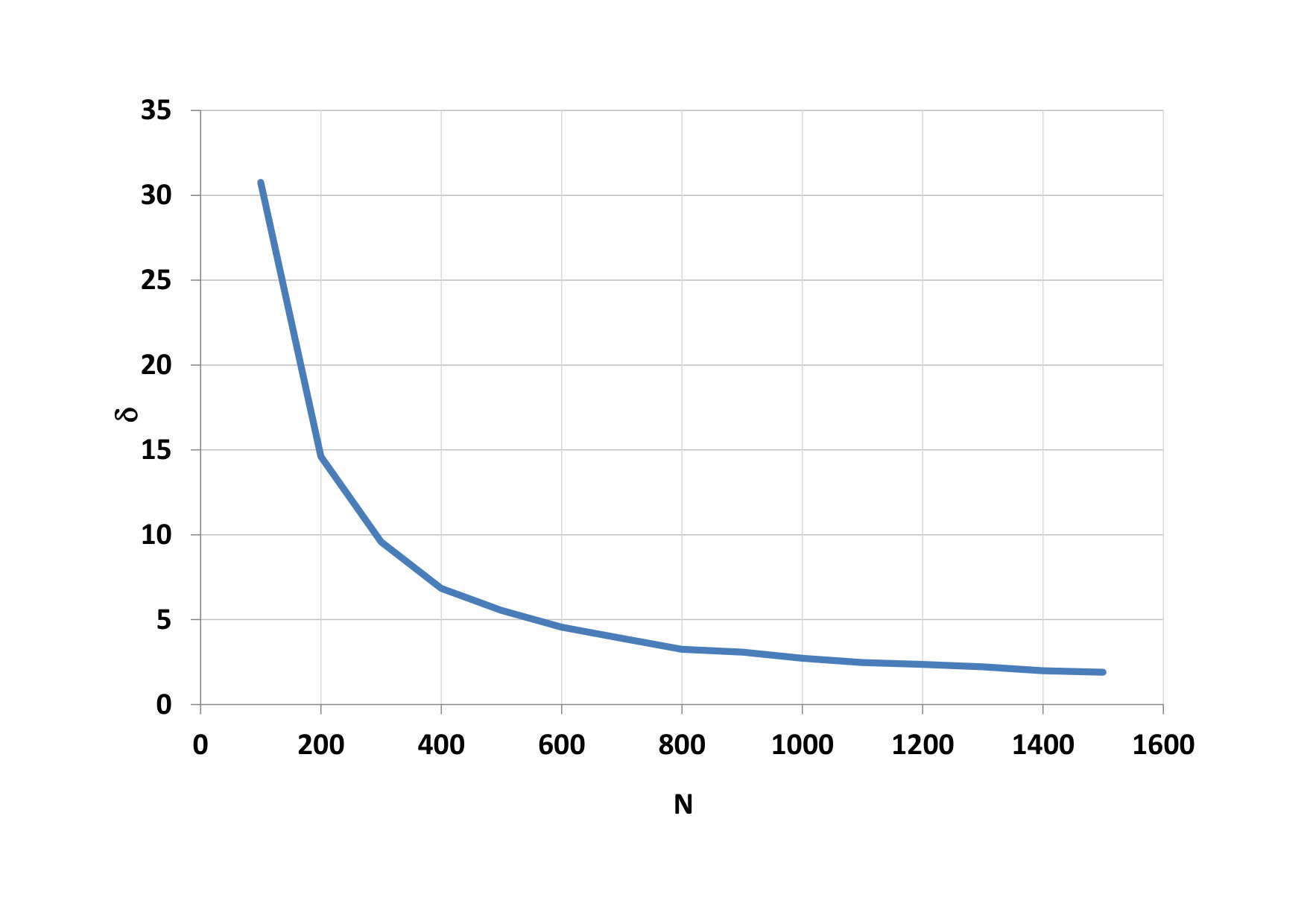 Figure 4
Figure 4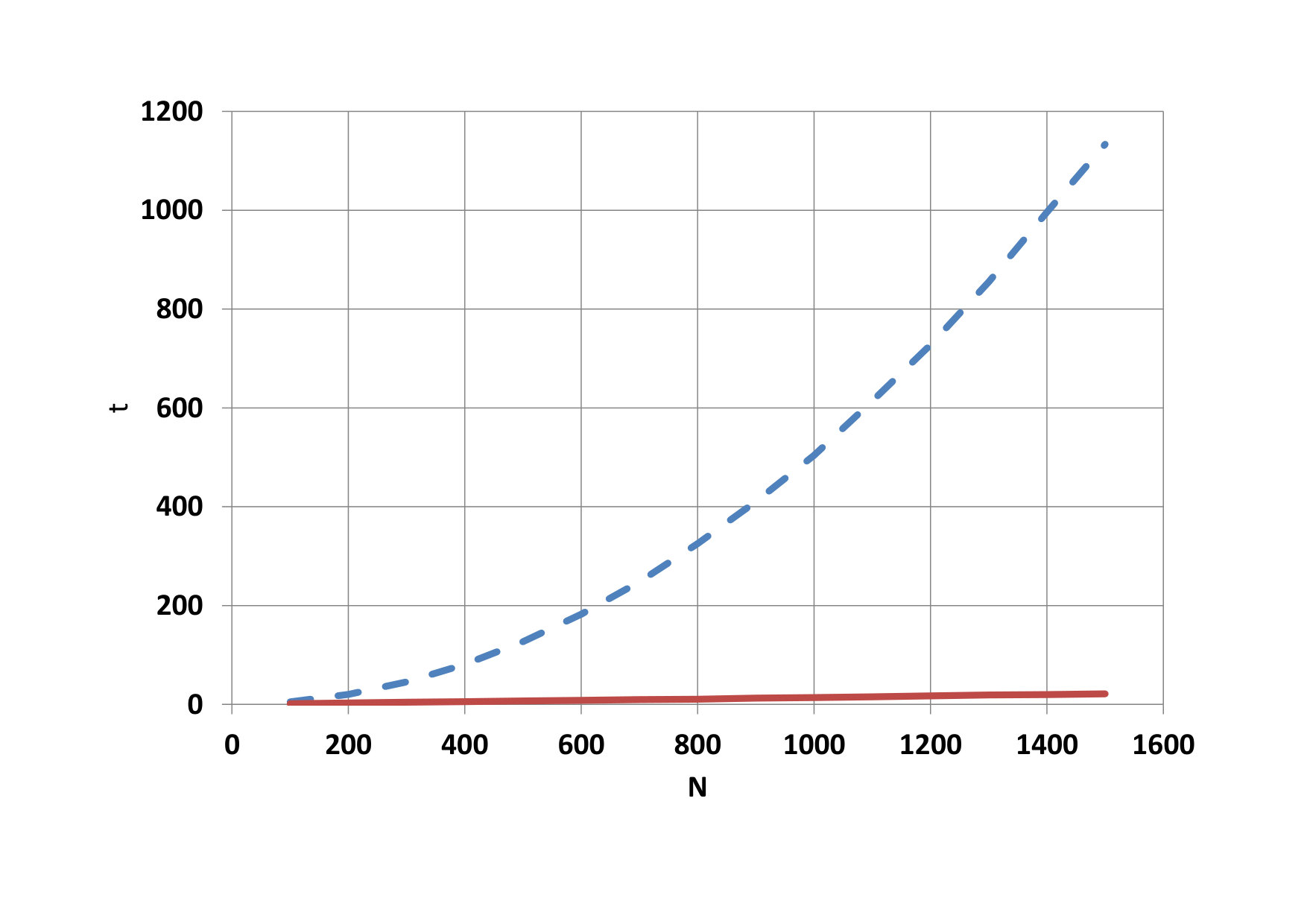 Figure 5
Figure 5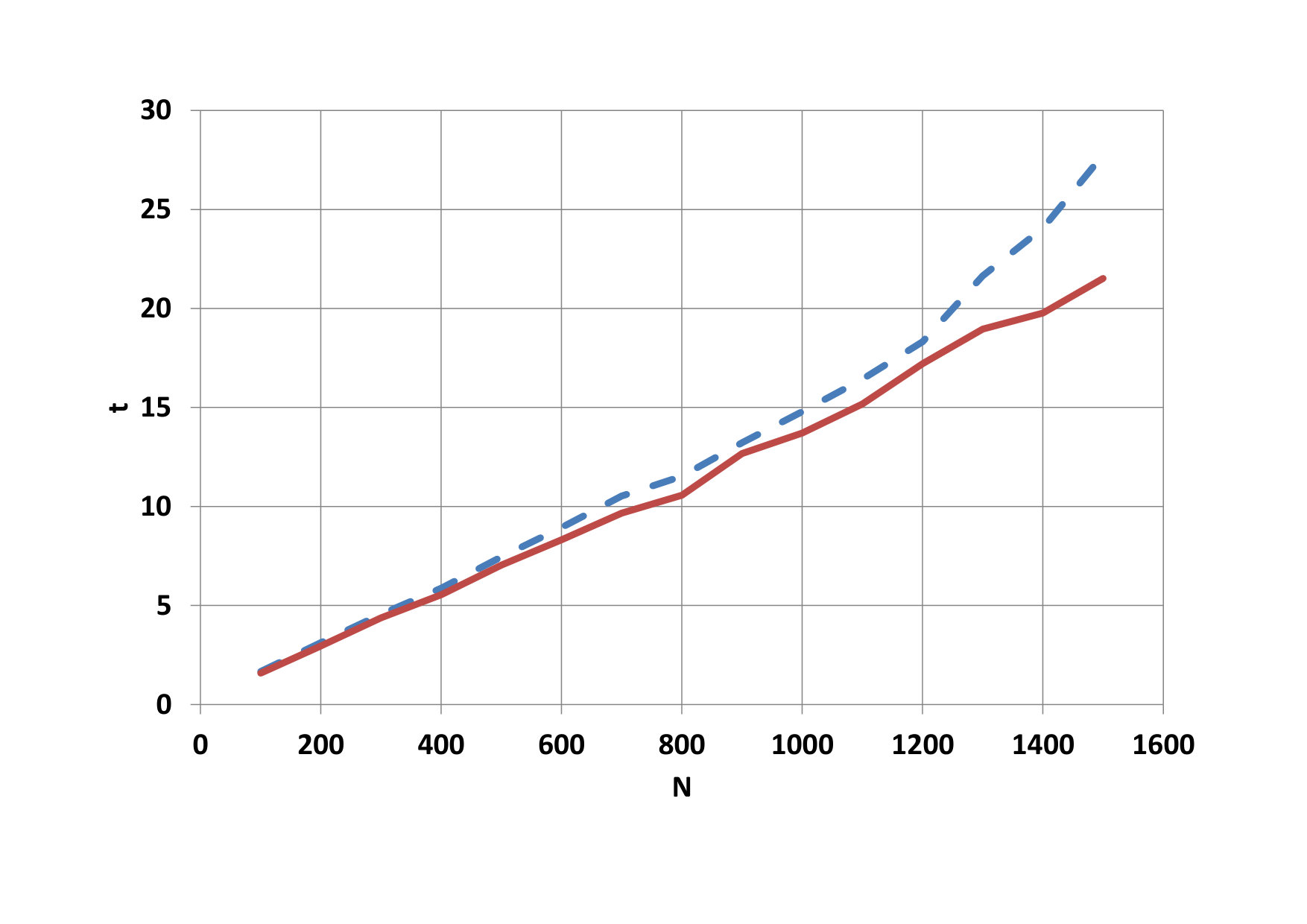 Figure 6
Figure 6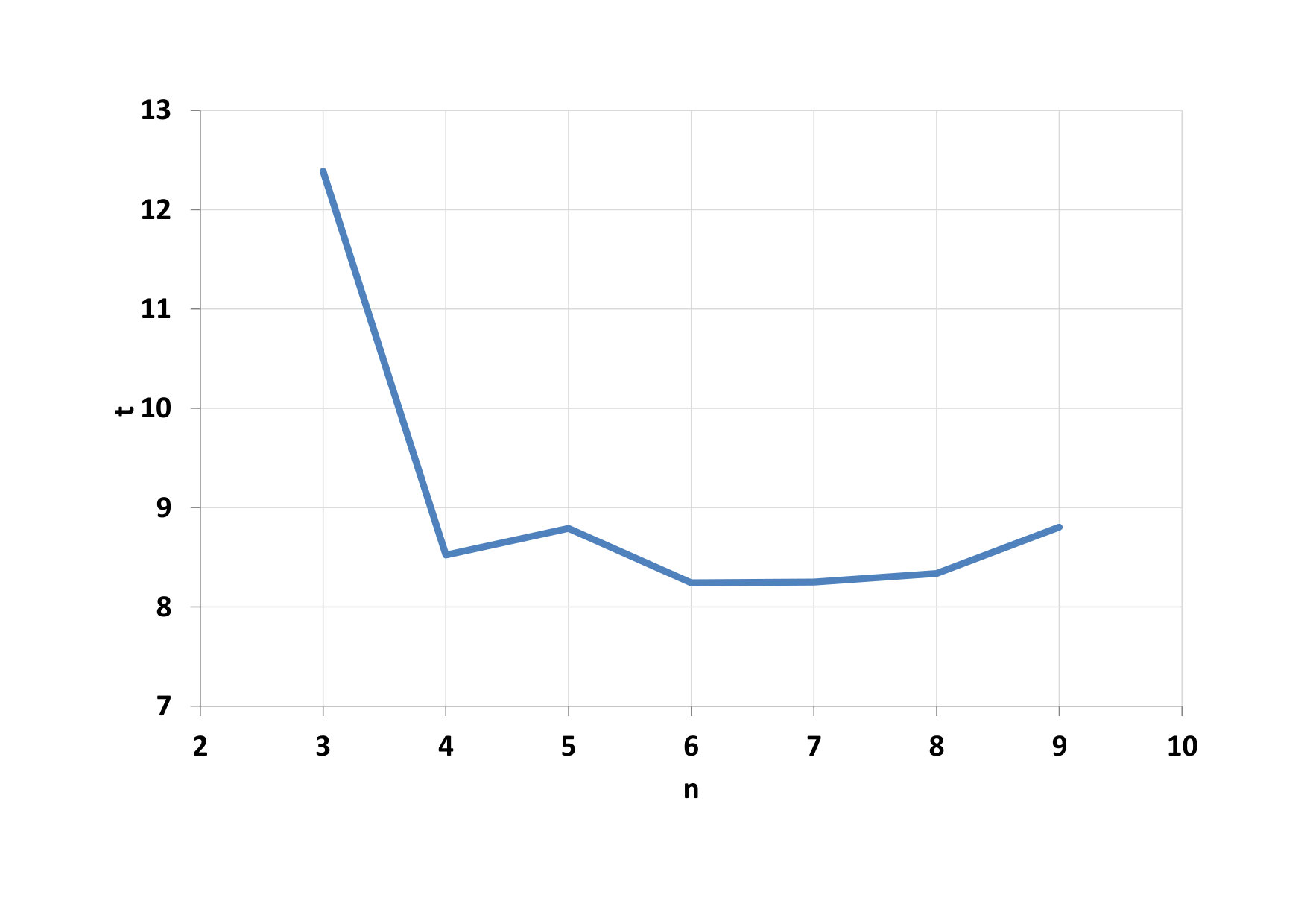 Figure 8
Figure 8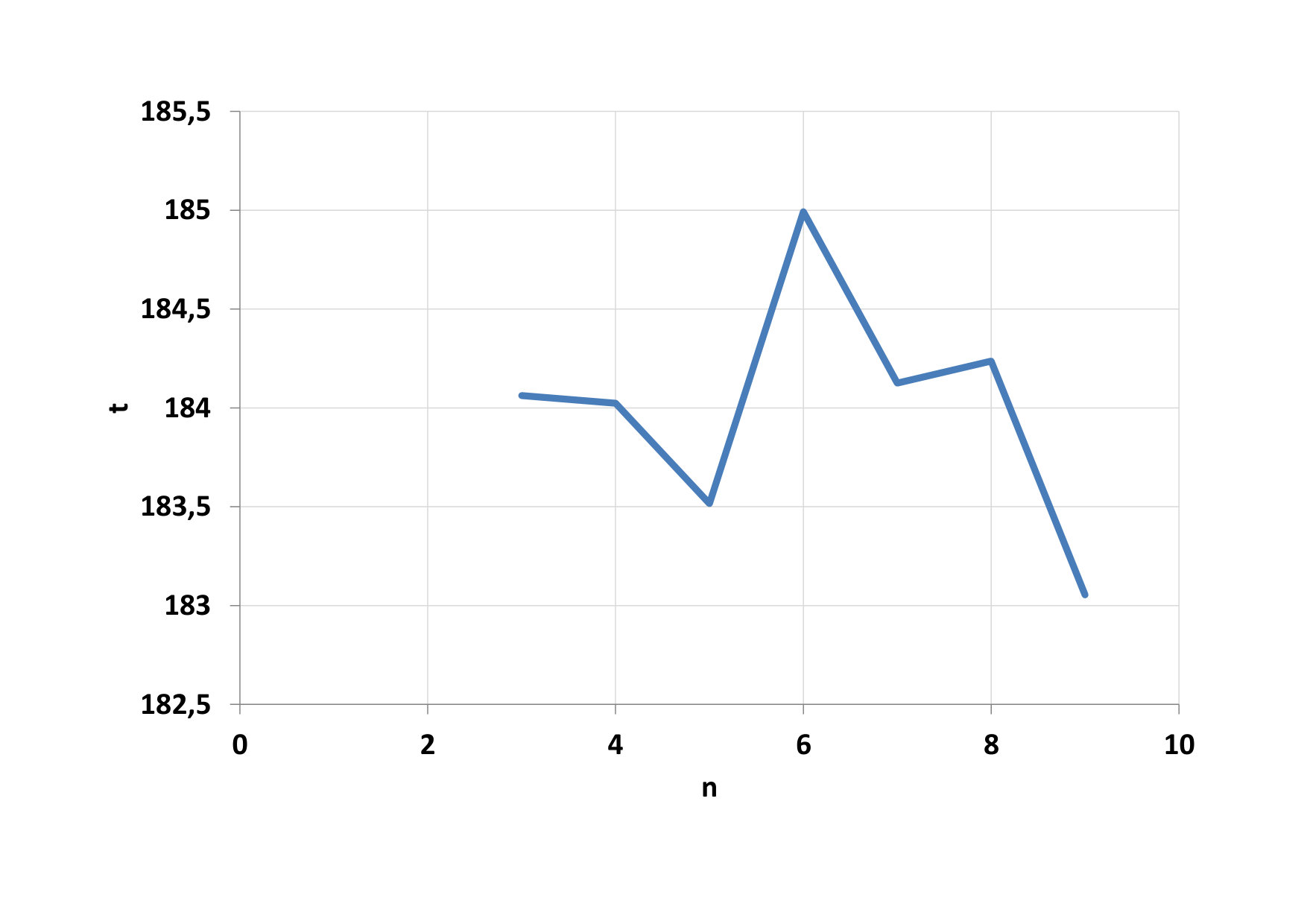 Figure 9
Figure 9| F | () | () | ||
|---|---|---|---|---|
| F1 | 112.575 | 2.372 | 4.479 | 0.110 |
| F2+F3 | 0.124 | 0.005 | 0.135 | 0.009 |
| F4 | 0.038 | 0.002 | 0.054 | 0.003 |
| 100 | 5.125 | 1.577 | 30.775 |
|---|---|---|---|
| 200 | 20.238 | 2.957 | 14.610 |
| 300 | 45.588 | 4.371 | 9.589 |
| 400 | 81.017 | 5.541 | 6.839 |
| 500 | 126.854 | 7.040 | 5.550 |
| 600 | 182.441 | 8.317 | 4.559 |
| 700 | 248.107 | 9.660 | 3.894 |
| 800 | 325.447 | 10.563 | 3.246 |
| 900 | 410.330 | 12.675 | 3.089 |
| 1000 | 503.784 | 13.706 | 2.721 |
| 1100 | 614.173 | 15.175 | 2.471 |
| 1200 | 727.594 | 17.212 | 2.366 |
| 1300 | 855.108 | 18.956 | 2.217 |
| 1400 | 996.005 | 19.767 | 1.985 |
| 1500 | 1133.698 | 21.522 | 1.898 |
| 100 | 1.993 | 1.042 | 52.279 |
|---|---|---|---|
| 200 | 7.417 | 1.776 | 23.944 |
| 300 | 16.605 | 2.504 | 15.077 |
| 400 | 29.462 | 3.374 | 11.453 |
| 500 | 46.049 | 4.115 | 8.936 |
| 600 | 68.389 | 4.829 | 7.061 |
| 700 | 90.600 | 5.571 | 6.149 |
| 800 | 118.330 | 6.162 | 5.207 |
| 900 | 150.655 | 7.276 | 4.830 |
| 1000 | 184.301 | 8.175 | 4.436 |
| 1100 | 223.147 | 9.035 | 4.049 |
| 1200 | 263.869 | 9.924 | 3.761 |
| 1300 | 313.163 | 10.501 | 3.353 |
| 1400 | 360.352 | 11.587 | 3.215 |
| 1500 | 412.590 | 12.434 | 3.014 |
| time | 2D | 3D | ||||
| CPU | pure GPU | total GPU | CPU | pure GPU | total GPU | |
| One Step | 112.732 | 4.532 | 4.668 | 184.349 | 8.649 | 8.786 |
| Per Particle | 0.05162 | 0.00207 | 0.00214 | 0.06584 | 0.00309 | 0.00314 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMethane Hydrates and Related Phenomena · Scientific Research and Discoveries · Advanced Physical and Chemical Molecular Interactions
Molecular dynamic simulation of water vapor interaction
with various types of pores using hybrid computing structures ††thanks: topic JINR LIT No. 05-6-1118-2014/2019, protocol No. 4596-6-17/19. HybriLIT resources.
V.V. Korenkov1, E.G. Nikonov1, M. Popovičová2
1*Joint Institute for Nuclear Research,
141980 Dubna, Moscow Region, Russia
email: [email protected], [email protected]
2University of Prešov,
str. Konštantinova 16, 080 01 Prešov, Slovakia
email: [email protected]*
Abstract
Theoretical and experimental investigations of water vapor interaction with porous materials are very needful for various fields of science and technology. Not only studies of the interaction of water vapor and porous material as a continuous medium, but also the study of the interaction of water vapor with individual pore is very important in these researches. Mathematical modelling occupies an important place in these investigations. Conventional approaches to solve problems of mathematical research of the processes of interaction of water vapor with individual pore are the following. The first approach is based on the use of diffusion equation for description of interaction of water vapor with a pore. It is so called macro approach. The second approach is based on various particle methods like, for example, molecular dynamics (MD). These methods essentially consider the micro-structure of the investigated system consisting of water vapor and a pore. This second approach can be called a micro approach.
At the macro level, the influence of the arrangement structure of individual pores on the processes of water vapor interaction with porous material as a continuous medium is studied. At the micro level, it is very interesting to investigate the dependence of the characteristics of the water vapor interaction with porous media on the geometry and dimensions of the individual pore. Both approaches require the most efficient calculation methods as far as possible with the current level of development of computational technologies. Usage of efficient calculation methods is necessary because the degree of approximation for simulating system is largely determined by the dimensionality of the system of equations being solved at every time step. Number of time steps is also quite large.
In this work, a study of efficiency of various implementations algorithms for MD simulation of water vapor interaction with individual pore is carried out. A great disadvantage of MD is its requirement of a relatively large computational effort and long time in simulations. These problems can be drastically reduced by parallel calculations. In this work we investigate dependence of time required for simulations on different parameters, like number of particles in the system, shape of pores, and so on. The results of parallel calculations are compared with the results obtained by serial calculations.
Keywords: porous media, molecular dynamics, macroscopic diffusion model, parallel calculations
1 Introduction
One of the most important problem in numerical simulation based on molecular dynamics or Monte-Carlo approach of many particle systems is the need to use huge computing resources to obtain more or less realistic simulation results. A system of water vapor and a pore is an example of such many particle systems. Theoretical and experimental investigations of water vapor interaction with porous materials are very needful for various fields of science and technology. Not only studies of the interaction of water vapor and porous material as a continuous medium, but also the study of the interaction of water vapor with individual pore is very important in these researches. Mathematical modelling occupies an important place in these investigations. Conventional approaches to solve problems of mathematical research of the processes of interaction of water vapor with individual pore are the following. The first approach is based on the use of diffusion equation for description of interaction of water vapor with a pore. It is so called macro approach. The second approach is based on various particle methods like, for example, molecular dynamics (MD). These methods essentially consider the micro-structure of the investigated system consisting of water vapor and a pore. This second approach can be called a micro approach.
At the macro level, the influence of the arrangement structure of individual pores on the processes of water vapor interaction with porous material as a continuous medium is studied. At the micro level, it is very interesting to investigate the dependence of the characteristics of the water vapor interaction with porous media on the geometry and dimensions of the individual pore. Both approaches require the most efficient calculation methods as far as possible with the current level of development of computational technologies. Usage of efficient calculation methods is necessary because the degree of approximation for simulating system is largely determined by the dimensionality of the system of equations being solved at every time step. Number of time steps is also quite large.
In this work, a study of efficiency of various implementations algorithms for MD simulation of water vapor interaction with individual pore is carried out. A great disadvantage of MD is its requirement of a relatively large computational effort and long time in simulations. These problems can be drastically reduced by parallel calculations. In this work we investigate dependence of time required for simulations on different parameters, like number of particles in the system, shape of pores, and so on. The results of parallel calculations are compared with the results obtained by serial calculations. Two-dimensional and three-dimensional models of the pore are used for comparative analysis of parallel and serial calculations.
2 Molecular dynamics model
In classical molecular dynamics, the behavior of an individual particle is described by the Newton equations of motion [Gould, 2005], which can be written in the following form
[TABLE]
where a particle number, , the total number of particles, particle mass, coordinates of position, the resultant of all forces acting on the particle. This resultant force has the following representation
[TABLE]
where the potential of particle interaction, a force caused by external fields. For a simulation of particle interaction, we use the Lennard-Jones potential [Lennard-Jones, 1924] with and eV. It is the most used to describe the evolution of water in liquid and saturated vapor form. Equations of motion (1) were integrated by Velocity Verlet method [Verlet, 1967]. Berendsen thermostat [Berendsen, 1984] is used for temperature calibration and control. The coefficient of the velocity recalculation at every time step depends on the so called ”rise time” of the thermostat which belongs to the interval . describes strength of the coupling of the system to a hypothetical heat bath. For increasing , the coupling weakens, i.e. it takes longer to achieve given temperature from current temperature The Berendsen algorithm is simple to implement and it is very efficient for reaching the desired temperature from far-from-equilibrium configurations.
Initial concentrations were obtained from the density of water vapor at the appropriate pressure and density at a given temperature using known tabulated data. The pressure in the pore was controlled using the formula based on virial equation [Frenkel and Smith, 2002].
[TABLE]
Here is the pore volume, is the doubled kinetic energy averaged over the ensemble, is the force between particles and at a distance .
3 Computational algorithm for molecular dynamic simulation
For molecular dynamic simulation we used the code written in CUDA C. The program does not require a lot of memory. We only keep co-ordinates, speeds and forces for each particle. One of the main problems of molecular dynamic simulation is a large number of particles and time steps. Therefore it is necessary to use parallel calculations. The code for our simulations was implemented on heterogeneous computing cluster HybriLIT.
The code contains four functions that are paralleled and which are performed on the GPU. This is a function for calculating the forces (i.e., acceleration) for individual particles, which calculates the interactions between all particles (F1). There are two functions to calculate new coordinates and speeds for each particle. We need two functions to calculate them because we need forces acting on particles at two different time moments(F2 and F3). Finally, we use the Berendsen thermostat in the program that runs parallel to the GPU too (F4).
We use natural parallelism for molecular dynamic simulations. The force calculations and velocity/position updates can be done simultaneously for all particles.
There are two basic ideas how to achieve parallelism. The goal in each is to divide computations evenly across the processors so as to extract maximum effect.
In the first class of methods a subgroup of particles is assigned to each processor. This method is called an particle-decomposition of the workload. The processor performs all calculations on its particles no matter where they move in the simulation domain.
The second group of methods is called a spatial decomposition of the workload. It means that parts of the physical simulation domain is assigned to each processor. Each processor only works with the particles in its subdomain.
Our program uses an particle-decomposition method. One command provides processing of a large amount of data that depends on how the block is defined in the program. The pseudo code for all four parallel functions is in Fig. 1 - 4
Other calculations are performed on the host. General scheme of the calculation algorithm for two- and three-dimensional molecular dynamic simulation is shown in Fig. 5.
In this paper we compare the temporal realization of these four functions on the GPU and the CPU. The total time of parallel computing consists of two parts, that is the time needed directly to calculate on the GPU (pure GPU time) and the time needed to complete these calculations on the CPU because some algorithms performed on the GPU must be completed by the CPU. In this work, total GPU time will indicate the sum of these two times.
4 2D molecular dynamic simulation
We consider the pore with dimensions , . The outer space in this micro-model reflects as a space right to the pore, see Fig. 6 (dashed line) which size, one can change by means of the parameter .
All sides of the outer space satisfy to the periodic boundary conditions. The left pore side reflects the inner molecules due to the boundary condition [NPP, arXiv:1709] but also provides the periodic boundary conditions for a part of outer space.
There are molecules of water vapor inside the pore which form saturated water vapor at temperature C and pressure at the time . The value of parameter means that the outer space volume for calculations is times larger that the pore volume. There are molecules of water vapor in outer space corresponding to 20% saturated water vapor. The integration step is .
First, we made several runs of our program with 69 blocks of 32 threads. Each implementation took 2000 time steps. We found that performing all the functions at every step in each run is the same with a small deviation. Time averages and deviations for each function, as well as the overall time of one step are shown in table 1.
For this reason, we will further consider that all program runs take an average implementation of time.
Furthermore, the total time of calculating the parallel portion of the code was examined, depending on the number of threads in the blocks (Fig. 7). We see that the minimum time has been reached for 128-threaded blocks (n = 7). Such a dependency pattern did not have all 4 functions that are executed in parallel. The main creator of this result was a function to calculate the potential (F1). The calculation time on CPU varies within the calculated standard deviation (Fig. 8). When comparing CPU and GPU calculations time, we can see that GPU calculations were performed on average 24 times faster than CPU calculations.
Finally, we studied the calculation time for both platforms, depending on the number of particles in the pore while maintaining the ratio of the density in the pores and in the outer area of 5 : 1. It was used blocks with 128 threads. The results can be seen in table 2.
On Fig.9, we can see as it grows advantage of parallel computing when the number of particles increases. Time needed for calculation on the CPU a total time on the GPU is compared on figure 10. The development of the GPU calculation time for blocks with different threads is shown on figure 11.
5 3D molecular dynamic simulation
In three-dimensional case[NPP, arXiv:1708] we made simulation for a pore in the shape of a prism of dimensions nm, nm, nm. Five walls are isolated and there is no exchange of particles with outer space. The sixth wall is open. The external environment is illustrated by a prism which is 9 times bigger than the pore. The big prism satisfies periodic boundary conditions. This means that the particles which pass through one wall return to the system through the opposite wall. Integration time step is ps and evolution time 65.3 ns. For our purposes, we will again perform only 2000 time steps.
Consequently, we have considered the following input data for the drying process: There are 1000 molecules of water vapor inside the pore which form saturated water vapor at temperature and pressure . There are 1800 molecules of water vapor in the outer area space corresponding to 20% saturated water vapor.
The simulation of this problem is solved using the CUDA C code according to the computational scheme on Fig. 5. Each of the 4 functions F1 - F4 is expanded to calculate the 3rd coordinate.
We first look at the dependence of the total computational time for parallel computing on the number of threads in the block, this dependence can be seen on Fig. 12. The minimum time has been reached for blocks with 64, 128 and 256 threads (n = 6, 7, 8). Again the main creator of this result was a function to calculate the potential (F1). GPU calculations were performed on average 21 times faster than CPU calculations. Development of computational time on the CPU is shown on Fig. 13.
Furthermore, the calculation time of both platforms was investigated depending on the number of particles in the pore. For 3D simulation, the same ratio of particles inside the pores and in outside was maintained like for 2D simulation. 128-threaded blocks were used for the calculations. The results are shown in Table 3.
The advantage of parallel calculations for the increasing number of particles is shown on Fig. 14. Comparison of CPU time and total GPU time is depicted on Fig. 15. The development of the GPU calculation time for blocks with different threads is shown on Fig. 16.
For both simulations, the average time required to calculate one step was also calculated. In order to compare the two simulations, we have converted this time to one particle. The results are shown in Table 4. The time required to calculate one step for one particle for 2D and 3D simulation on GPU in both cases (pure GPU time and total GPU time) especially pure GPU time is 2 : 3. CPU time does not keep this ratio.
6 Conclusions
As our investigations showed for both cases of 2D and 3D simulation, when paralleling the computations, there are some optimal value of number of threads in blocks such that the computation time becomes minimal in comparison with other values of this number of threads. In addition, it should be noted that, when parallelizing, the cost ratio of the computation time per particle for 2D and 3D modeling is equal with high precision.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1[Berendsen, 1984] H.J.C.Berendsen, J.P.M.Postma, W.F.van Gunsteren, A.Di Nola, J.R.Haak, Molecular dynamics with coupling to an external bath. // J. Chem. Phys. — 1984, — vol. 81, — P. 3684–3690.
- 2[Gould, 2005] Gould H., Tobochnik J., Christian W., An Introduction to Computer Simulation Methods, Chapter 8. Third edition, 2005, pp. 267-268.
- 3[Frenkel and Smith, 2002] Frenkel D., Smith B., Understanding molecular simulation : from algorithms to applications. Second edition, Academic Press, 2006, 658 pp.
- 4[Lennard-Jones, 1924] J. E. Lennard-Jones, On the Determination of Molecular Fields. // Proc. Roy. Soc. — 1924, — vol. A 106, — P. 463 -477.
- 5[NPP, ar Xiv:1709] Nikonov E.G., Pavluš M., Popovičová M., 2D microscopic and macroscopic simulation of water and porous material interaction. — 2017, — ar Xiv:1709.05878 [physics.flu-dyn]
- 6[NPP, ar Xiv:1708] Nikonov E.G., Pavluš M., Popovičová M., Molecular dynamic simulation of water vapor interaction with blind pore of dead-end and saccate type. — 2017, — ar Xiv:1708.06216 [physics.flu-dyn]
- 7[Verlet, 1967] Verlet L., Computer ’experiments’ on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. // Phys. Rev. — 1967, — vol. 159, — p. 98–103.