Anticipating acene-based chromophore spectra with molecular orbital arguments
Timothy J. H. Hele, Eric G. Fuemmeler, Samuel N. Sanders, Elango, Kumarasamy, Matthew Y. Sfeir, Luis M. Campos, Nandini Ananth

TL;DR
This study combines synthesis, electronic structure theory, and spectral analysis to explain and predict the absorption spectra of acene-based chromophores, revealing a design principle for creating highly absorbent molecules.
Contribution
It introduces a molecular orbital-based rule to predict absorption features in acene dimers, explaining a long-standing spectral phenomenon and guiding chromophore design.
Findings
Charge-transfer excitations borrow intensity from UV states.
The bonding geometry determines the presence of additional absorption.
The rule accurately predicts spectra of various acene derivatives.
Abstract
Recent synthetic studies on the organic molecules tetracene and pentacene have found certain dimers and oligomers to exhibit an intense absorption in the visible region of the spectrum which is not present in the monomer or many previously-studied dimers. In this article we combine experimental synthesis with electronic structure theory and spectral computation to show that this absorption arises from an otherwise dark charge-transfer excitation 'borrowing intensity' from an intense UV excitation. Further, by characterizing the role of relevant monomer molecular orbitals, we arrive at a design principle that allows us to predict the presence or absence of an additional absorption based on the bonding geometry of the dimer. We find this rule correctly explains the spectra of a wide range of acene derivatives and solves an unexplained structure-spectrum phenomenon first observed seventy…
Click any figure to enlarge with its caption.
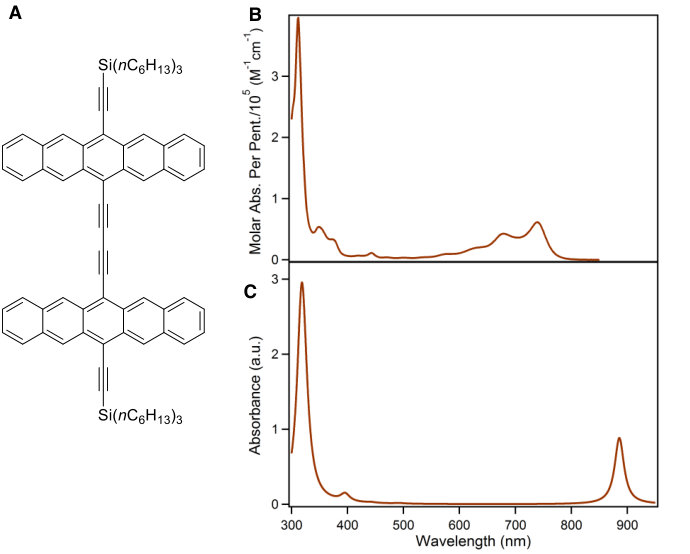 Figure 1
Figure 1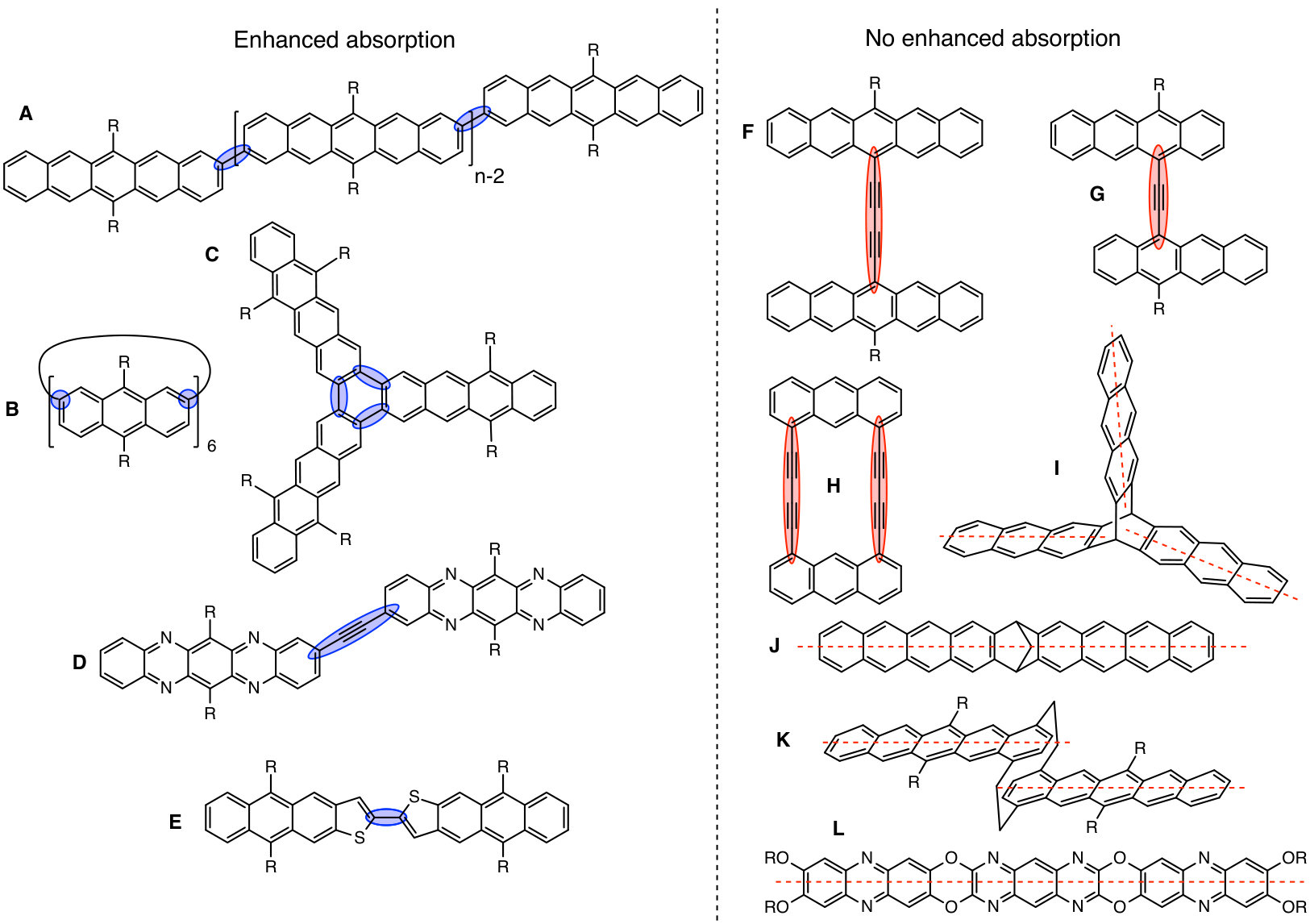 Figure 2
Figure 2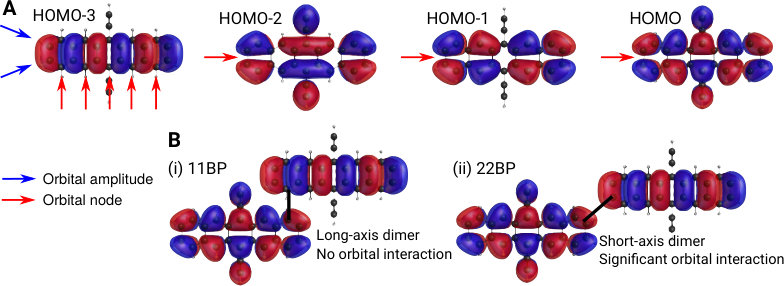 Figure 3
Figure 3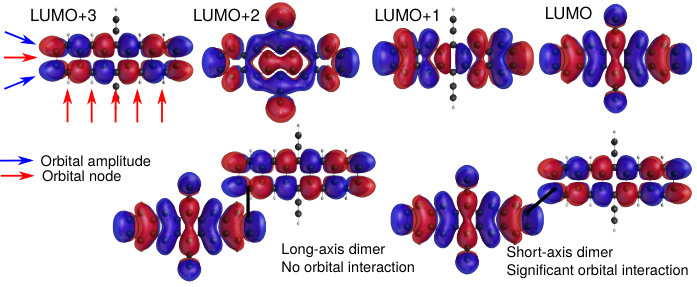 Figure 4
Figure 4 Figure 5
Figure 5 Figure 6
Figure 6 Figure 7
Figure 7 Figure 8
Figure 8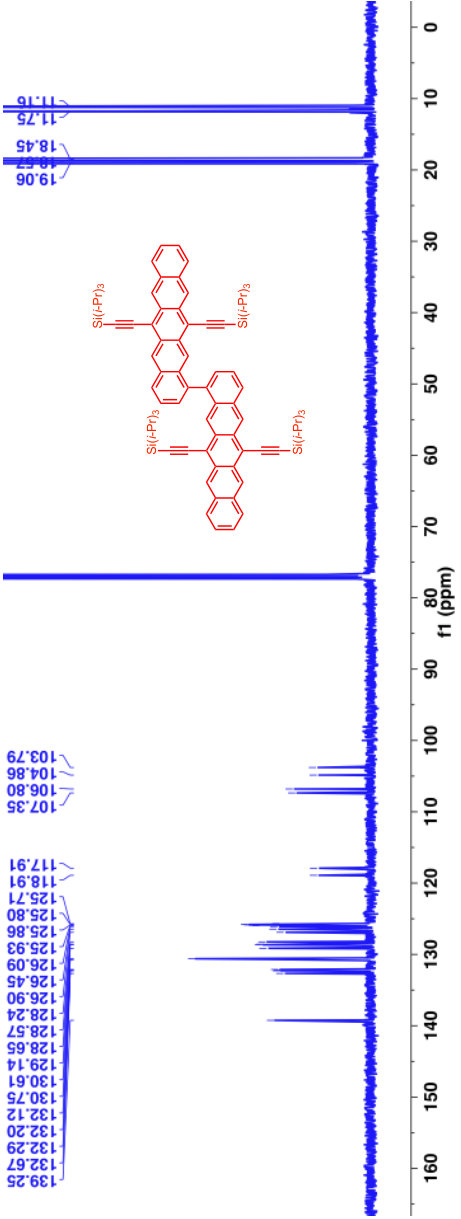 Figure 9
Figure 9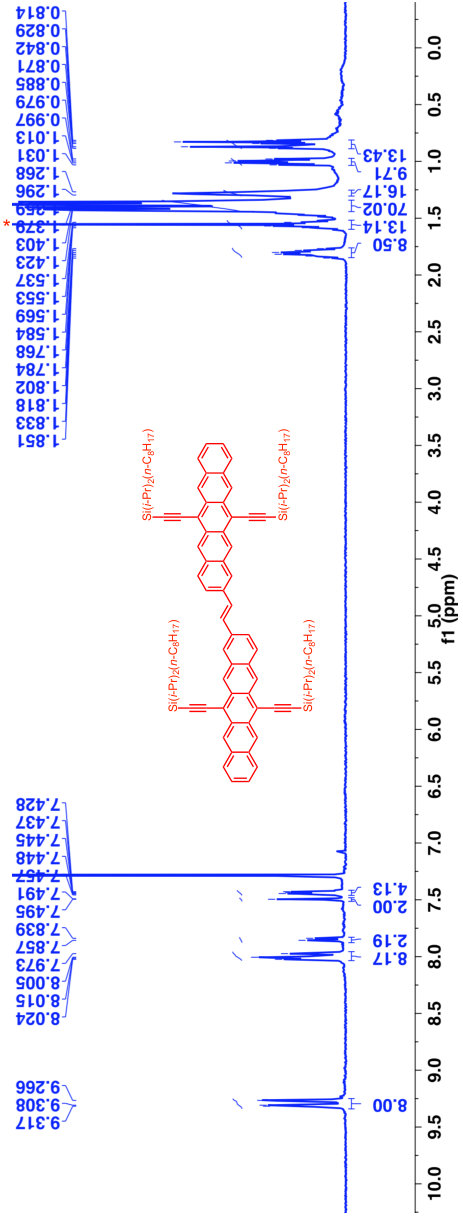 Figure 10
Figure 10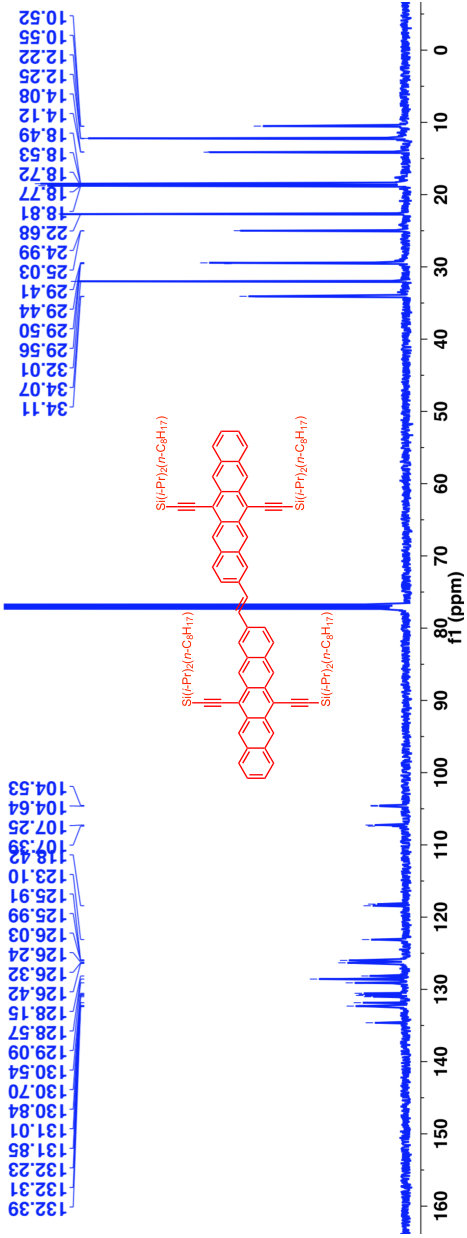 Figure 11
Figure 11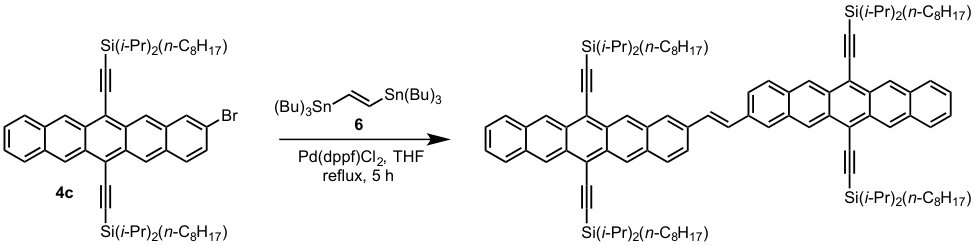 Figure 12
Figure 12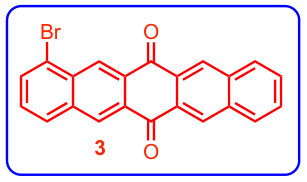 Figure 13
Figure 13 Figure 14
Figure 14 Figure 15
Figure 15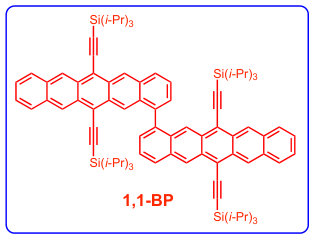 Figure 16
Figure 16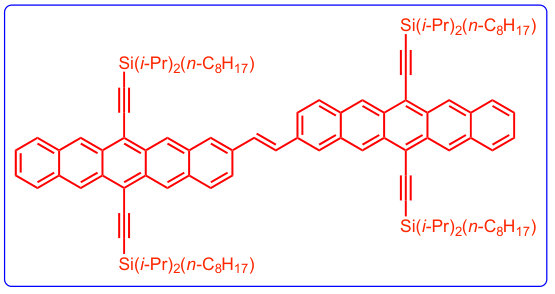 Figure 17
Figure 17 Figure 18
Figure 18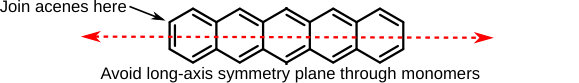 Figure 19
Figure 19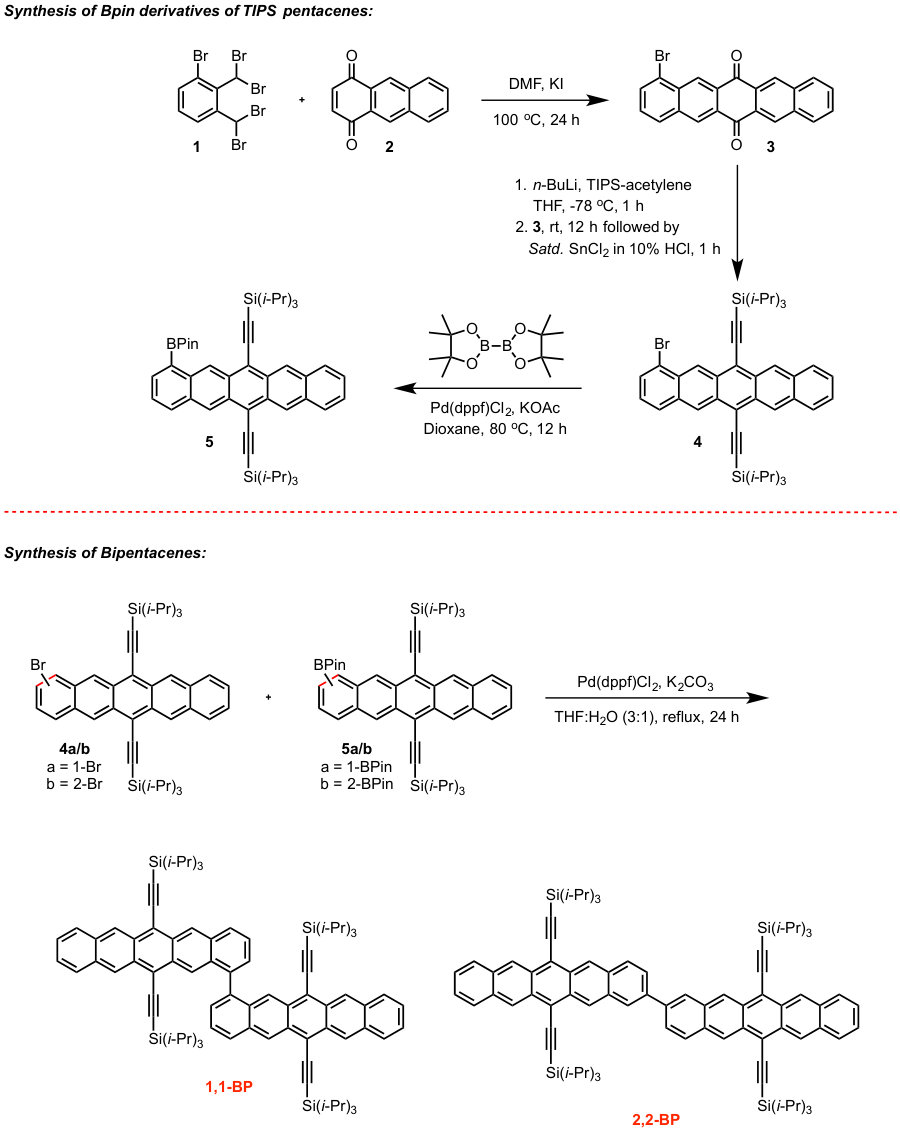 Figure 20
Figure 20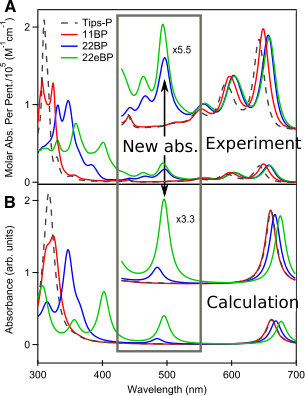 Figure 21
Figure 21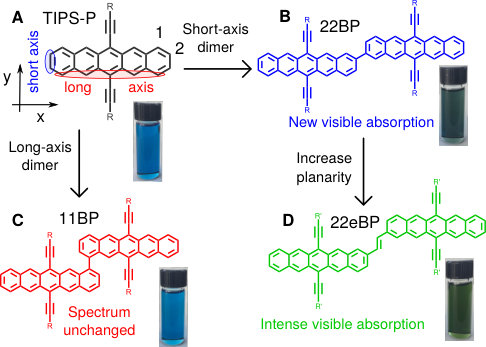 Figure 22
Figure 22Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Anticipating acene-based chromophore spectra with molecular orbital arguments
Timothy J. H. Hele
Cavendish Laboratory, JJ Thomson Avenue, Cambridge University, CB3 0HE, UK
Eric G. Fuemmeler
Baker Laboratory, 259 East Avenue, Cornell University, Ithaca, NY 14850, USA
Samuel N. Sanders
Elango Kumarasamy
Department of Chemistry, Columbia University, New York, NY 10027, USA
Matthew Y. Sfeir
Center for Functional Nanomaterials, Brookhaven National Laboratory, Upton, NY 11973, USA
Luis M. Campos
Department of Chemistry, Columbia University, New York, NY 10027, USA
Nandini Ananth
Baker Laboratory, 259 East Avenue, Cornell University, Ithaca, NY 14850, USA
Abstract
Recent synthetic studies on the organic molecules tetracene and pentacene have found certain dimers and oligomers to exhibit an intense absorption in the visible region of the spectrum which is not present in the monomer or many previously-studied dimers. In this article we combine experimental synthesis with electronic structure theory and spectral computation to show that this absorption arises from an otherwise dark charge-transfer excitation ‘borrowing intensity’ from an intense UV excitation. Further, by characterizing the role of relevant monomer molecular orbitals, we arrive at a design principle that allows us to predict the presence or absence of an additional absorption based on the bonding geometry of the dimer. We find this rule correctly explains the spectra of a wide range of acene derivatives and solves an unexplained structure-spectrum phenomenon first observed seventy years ago. These results pave the way for the design of highly absorbent chromophores with applications ranging from photovoltaics to liquid crystals.
keywords:
Chromophore design, UV-vis spectra, Acenes, Electronic Structure Theory
\SectionNumbersOn\abbreviations
1 Introduction
Investigation of the electronic structure and spectra of organic molecules has a long history, dating back to the molecular orbital theories of Hückel1 and the oscillator strength sum rules of Thomas, Reiche and Kuhn (TRK)2, 3, 4. These have been followed by development and application of more sophisticated electronic structure methods5, 6, 7, 8, 9, 10, 11, 12, 13, 14 and the formulation of structure-spectrum design principles such as Kasha’s point-dipole model15, crystallochromy16, and the tuning of absorption frequencies (color) by alteration of orbital energy gaps17. Despite this considerable progress, there remains a need for more systematic design rules for the creation of absorbent and tunable chromophores18, 19, 20.
More recently, there has been a surge of interest in organic chromophores for the development of efficient photovoltaics21, 22. In particular, acenes such as tetracene and pentacene possess the unusual and useful ability to undergo singlet fission23, 24, 25, 26 which, by splitting one high-energy exciton into two low-energy triplets, has the potential to substantially increase the efficiency of organic solar cells.27, 28 This has led to the synthesis and characterization of a large range of dimers and oligomers of tetracene and pentacene, some of which exhibit an intense visible absorption in addition to the monomer transition29, 25, 30, whereas others do not31. This additional absorption occurs without any significant change in the intensity or frequency of the lowest-energy monomer () excitation, which is preserved in all these dimers29, 25, 30, 31. Examining the literature, we find that an interesting dependence of UV-vis spectra on the bonding geometry of acene dimers was first observed in 1948 for dimers of naphthalene (where all the transitions are in the UV),32 and has since been observed in many other acene derivatives 33, 34, 35, 36. Previous computational investigations have assigned this to a transition 33, 37 but there has been no clear explanation of its presence in some acene derivatives 32, 33, 34, 35, 36 and absence in others 38, 39, 40, 41, 42, 43, 44, 45.
While initially a curiosity, the ability to synthesize acene derivatives that exhibit enhanced visible absorptions may lead to organic photovoltaic materials with increased efficiency. In theory, using a more absorbent chromophore would allow for a decrease in the thickness of a cell while still absorbing the same proportion of solar radiation thereby reducing cost and increasing flexibility. The thinner cell would also allow more diffusing excitons to be extracted as current, increasing efficiency22, 46. There is, consequently, a theoretical and practical need to uncover the origin of this new absorption for the design of novel acene-derivative based materials.
In this article we present a joint experimental-theoretical investigation that finds the origin of this new visible absorption is ‘intensity borrowing’47 from an intense UV absorption. The presence or absence of intensity borrowing in dimers can be deduced by examining whether the relevant monomer molecular orbitals have amplitude on the carbon atoms through which the monomers are bonded. We find that the resulting design rule can be used to predict the absorption spectra of a very wide range of acene derivatives, including oligomers with unusual bonding geometries and heteroatom-substituted derivatives.
We begin by presenting experimental and computed acene spectra in section 2, finding that Pople-Parr-Pariser (PPP) theory5, 6, 7, 8 correctly predicts the presence or absence of the extra absorption and its approximate intensity. To explain why this additional absorption is observed in some dimers and not others, in section 3, we use intensity borrowing perturbation theory and PPP theory to analyze the relevant monomer molecular orbitals. In section 4 we apply this analysis to bipentacenes, explaining the origin of the new absorption in some dimers and its absence in others, and we formulate a general design rule for acene derivatives. We show that this design rule correctly predicts the presence or absence of an additional absorption in a large and diverse range of dimers, trimers, and oligomers in section 5. Conclusions are presented in section 6.
2 Acene dimer spectra
The spectra of acenes is a much-studied area (see, for example, Refs. 48, 8, 49, 10, 40, 50, 51, 52, 13, 14, 53, 54, 55, 10, 56, 12, 57, 58, 9) and here we focus on the particular case of covalently-linked dimers and oligomers of acenes. Although we find our results to be widely applicable, we present them in the context of Bis(triisopropylsilylethynyl)pentacene (TIPS-pentacene, Fig. Fig. 1A), a molecule of interest for its extensive applications in photovoltaics 24, 37, 25, 59, 38.
2.1 Experimental spectra
We start by comparing the experimental spectra of TIPS-pentacene and a recently-reported dimer, 2,2-bipentacene (22BP)25, 59, with molecules shown in Fig. 1 and spectra in Fig. 2. The spectra of TIPS-pentacene and 22BP have the familiar features of an acene spectrum8, 12, 53: a weak -polarized transition at low frequencies (coordinates defined in Fig. 1A), accompanied by a vibrational stretching progression and a very intense absorption in the near UV (310-350 nm). We note two distinct differences between the spectrum of 22BP and the monomer. First, 22BP exhibits a visible absorption around 500nm with its own vibrational stretching progression. Second, although the lowest energy transition () is unaffected by dimerization, the intensity of the UV absorption decreases and red-shifts. The extinction coefficients in Fig. 2 are plotted per pentacene to show that, in the visible, the 2, dimer is more absorbent than the sum of its parts and not simply an artifact of the chromophore being twice as large.
A previous study focusing on the singlet fission properties of these molecules59 considered bonding bulking phenyl groups around the inter-pentacene bond, finding that the size of the new absorption decreased as the twist angle between the two chromophores increased. Similarly, bonding a succession of benzene groups as linkers between the two pentacenes causes the new absorption to decrease in intensity. To investigate the origin of this visible absorption further, we synthesized two more pentacene derivatives (details in the supporting information). We make 1,1’-bipentacene (11BP, Fig. 1C) finding its spectrum in Fig. 2 to be qualitatively identical to the TIPS-P monomer spectrum in the visible, and with a splitting of the high-energy absorption in the UV. We also synthesize a planar analogue of 22BP, 2,2’-ethene-linked bipentacene (22eBP, Fig. 1D) by linking the monomers with an alkene bond, where the alkene bond can be loosely interpreted as a conjugated ‘wire’ between two chromophores. We find that 22eBP exhibits the most intense absorption in the visible region of the spectrum among the bipentacenes considered here.
2.2 Computed spectra
Since these are very large chromophores, we employ Pariser-Parr-Pople (PPP) theory 8, 5, 60, 61, 62 to calculate dimer spectra. PPP theory was developed for the calculation of low-lying excited electronic states of conjugated systems8, 5, and its similarity to simple molecular orbital (MO) theories such as Hückel theory allows for a chemically intuitive interpretation of results 60, 61. Unlike Hückel theory, however, PPP theory includes two-electron interactions (within the neglect of differential overlap (NDO) approximation) such that it correctly accounts for Coulomb and exchange interactions which are required to give accurate excited-state energies 7. Computational details are given in the SI where we also benchmark our monomer calculations against high-level multiconfiguational calculations, confirming the suitability of PPP theory.
Examining Fig. 2, we see that the PPP spectra are in good agreement with experimental spectra (the computed energies are not shifted to agree with experiment) and correctly predicts a visible absorption in 22BP and 22eBP that is not present in either 11BP or the monomer. While the visible absorption in 22BP is somewhat weaker and slightly blue-shifted compared to experiment, in 22eBP it is accurately captured, both in terms of absorption frequency and relative intensity. We also see that PPP correctly predicts a Davydov splitting of the 11BP absorption (seen as a peak with a shoulder around 320nm), and a diminution in the height of the UV peaks of 22BP and 22eBP, although their relative intensities are inaccurate.
From our PPP calculations (verified by high-level calculations reported in the SI), we attribute the dimer excitation around 650nm to a predominantly HOMOLUMO transition. The intense UV peak arises from an in-phase combination of HOMO–3 to LUMO and HOMO to LUMO+3, whose corresponding out-of-phase combination is dark in PPP theory and is seen as a weak absorption at 440 nm in experiment. Excitations from the HOMO–1 to LUMO and HOMO to LUMO+1 are dipole forbidden, and the excitations from the HOMO–2 to the LUMO and the HOMO to the LUMO+2 are predicted by PPP theory to be -polarized, giving a dark out-of-phase state and a bright in-phase state at 358nm and 366nm respectively. The bright state probably corresponds to a shoulder in the intense UV absorption seen experimentally around 350nm.
3 Elucidating dimer spectra
We investigate the origin of the visible absorption in 22BP and 22eBP using intensity borrowing perturbation theory47, where we contruct a zeroth-order set of states (corresponding to non-interacting monomers), and examine the perturbation (if any) introduced by dimerization by bonding at different positions.
We begin by noting that textbook arguments63 fail to explain the variation in our dimer spectra. Conventional symmetry-breaking arguments fail to explain the extra absorption since both 11BP and 22BP are and the more symmetric 22eBP has the largest absorption (see Fig. 2). Similarly, a particle-in-a-box model63 predicts an intense HOMO to LUMO transition, which is inaccurate even for the monomer (Fig. 2), and it also predicts that the lowest-energy transition redshifts and grows in intensity upon enlarging the molecule, which is not observed. Furthermore, solvatochromism cannot explain the extra absorption, since alternant hydrocarbons64 such as acenes have no ground-state dipole or multipole. Planarity arguments could also be posited, since the overlap of the systems of the two monomers would be expected to increase with planarity, and 11BP is more twisted (nearly perpendicular) than 22BP (37*∘* dihedral angle59) and planar 22eBP. However, by linking pentacenes via an alkyne linker, a planar 6,6’-bipentacene has been synthesized38 and shows no extra absorption in the visible (SI Fig. 1). Consequently, planarity is advantageous but not sufficient.
More sophisticated theories of chromophore interaction include Kasha’s point-dipole model15, which can be used to explain the spectra of some acene derivatives65, 66. Kasha’s model requires two bright excitations to interact, and can therefore explain the splitting of the high-energy absorption seen in the UV spectrum of 11BP, but not the emergence of a new absorption in a region of the spectrum where there was previously none. The new dimer absorption cannot result from the monomer transition around 650nm splitting, since that would result in two absorption peaks with combined intensity equal to that of the monomer, whereas the dimer spectra of 22BP and 22eBP are more absorbent in the visible than the sum of their parts (see Fig. 2).
However, intensity borrowing perturbation theory47 is promising here, as it describes how dark transitions can ‘borrow intensity’ from bright states if they are coupled through a perturbation. Since oscillator strength is conserved by the TRK sum rule2, 3, 4, 63, the resulting new absorption in the spectrum is accompanied by a bright transition losing intensity. This is in keeping with our previous observation on the spectra of 22BP/22eBP where the appearance of a new visible absorption at 500nm is accompanied by a decrease in intensity of the UV peak. We also find that a previous computational study on 22BP which investigated its singlet fission properties and not the linear absorption spectrum, suggested the existence of a charge-transfer state around 440nm59. A reasonable hypothesis, is then, that a transition in the visible, possibly this CT excitation, is ‘borrowing intensity’ from the UV in certain dimers like 22BP and 22eBP but not in 11BP.
3.1 Intensity borrowing perturbation theory and PPP
We develop a theory to describe the interactions between the monomer UV excitations and the low-energy CT excitation in a dimer by combining intensity borrowing perturbation theory47 and PPP theory. We also characterize the role that dimer bonding geometry plays in modulating the strength of this interaction and, therefore, the appearance (or not) of an additional peak in the absorption spectrum.
We first formally define our system in the language of perturbation theory. Similar to Kasha15 and the ideas of symmetry-adapted perturbation theory67, for two monomers and , the Hamiltonian is
[TABLE]
where and are the Hamiltonian operators of the monomers at infinite separation and is the intermonomer perturbation. Kasha approximated as a dipole-dipole interaction and assumed all excitations were intra-monomer15, but here we make no a priori assumptions about the nature of the excitations nor the functional form of . Instead, we choose to describe the overall system using PPP theory5, 6, 7, 8 which we have already shown provides an accurate description of the electronic structure of these molecules.
To define the monomer Hamiltonians within PPP theory,5, 6, 7, 8, 61, 68 we let be the set of atoms on monomer such that
[TABLE]
and likewise for monomer , where is the number operator for electrons on atom ,
[TABLE]
where , and , are the creation and annihilation operators respectively for an electron of spin on atom . is the local chemical potential, which is 1 for a carbon atom. is the on-site energy, which for a purely hydrocarbon chromophore we can set to zero without affecting the energies of excited states. is the hopping term, where indicates the summation is over nearest neighbor pairs. is the on-site (Hubbard) repulsion, and is the parameterized repulsion between an electron on atom and an electron on atom (for details see SI).
Using the definition of the PPP Hamiltonian for an arbitrary chromophore5, 6, 7, 8, 61, 68 and Eq. (2), we define the intermonomer perturbation as
[TABLE]
Because the two chromophores are noninteracting at infinite separation, we can solve mean-field variants (Fock operators) of and separately for their corresponding molecular orbitals. The monomers are identical, allowing us to choose degenerate orbitals that are localized entirely on monomer or monomer . These molecular orbitals are denoted or where the subscript indicates the MO of monomer or . In accordance with convention8 bonding orbitals (those occupied in the ground state) are numbered from the HOMO downwards and antibonding orbitals from the LUMO upwards. As noted by Pariser8, care must be taken to be consistent with the relative signs of orbitals in order for the Coulson-Rushbrooke theorem64 to be easily applied. For the perturbation analysis of the dimers considered here we choose every monomer orbital to have the same sign on the atom through which it is joined to the other monomer.
We are interested in linear absorption spectra and the dipole moment is a one-electron operator, so we consider only singly excited states in keeping with the original formulation of PPP. We denote single excitations where and are either or . We only consider singlet spin-adapted configurations69 as triplet excitations are dark for hydrocarbons with minimal spin-orbit coupling. There are two types of single excitations. First, when , or , are intramolecular, local (Frenkel) excitations27, 57, 59, 70 that we denote as 70 . For alternant hydrocarbons64, 5, 8 such as the molecules considered in this article, and are degenerate and we therefore define ‘plus’ and ‘minus’ excitations8
[TABLE]
where only ‘plus’ excitations have nonzero transition dipole moment from the ground state.8
The second type of excitation (when ), or , are intermolecular, charge-transfer (CT) excitations57, 27, 70 which we denote . We appreciate that there are varying definitions and nomenclature for CT excitations in the literature27, 70, 71, 72, 73, and here the definition corresponds to removing an electron from an orbital localized entirely on one monomer and placing it in an orbital localized on another monomer. Using the definition of the dipole moment in PPP theory8 and that orbitals on different monomers are spatially disjoint, we note that the CT excitations defined here are always dark, .
Our zeroth order eigenstates of interest are thus the and states along with the ground state, . The only bright states at zeroth order will be the PPP ‘plus’ Frenkel excitations, .
3.2 First order perturbation
Having obtained zeroth-order eigenstates of , we now consider how they are mixed by the perturbation which occurs when the two monomer are covalently linked, and how this alters the linear absorption spectrum. Following standard perturbation theory63, we form the ‘good’ degenerate eigenstates of as linear combinations of excitations which we denote and in accordance with the irreducible representations (irreps) of the point group.63 For Frenkel excitations we have
[TABLE]
and for CT
[TABLE]
which is similar to the linear combinations used in (for example) Kasha exciton theory15, that considers only the dipole-dipole interactions of Frenkel excitations. Note that in certain cases of degeneracy further linear combinations may be required but we do not find this necessary in what follows.
Since the perturbation is symmetric under all the symmetry operations of the dimer, only like irreps can mix. We are therefore interested in the UV Frenkel excitations and mixing with the dark charge-transfer excitations and respectively. Using standard electronic structure theory algebra69 we find
[TABLE]
where is an element of the one-electron, hopping matrix in the molecular orbital basis. Note that due to the NDO approximation present within PPP theory, there are no two-electron terms in Eq. (9). Equation 8 shows that cannot borrow intensity from (to first order), and we therefore need only consider the corresponding excitations. This result is particularly convenient for planar or near-planar structures such as 22BP and 22eBP where dipole moment arguments show that only excitations of symmetry are likely to have significant oscillator strength.
To quantify the extent of mixing, we further simplify the right hand side of Eq. (9). Using the Coulson-Rushbrooke theorem64 and the definition of the sign of monomer orbitals (see above) we find that and . We also find from the symmetry of the dimer that and . We can then evaluate the one unique matrix element to obtain
[TABLE]
where represent the expansion coefficients for the monomer orbitals in the atomic orbital basis, . Due to the nearest-neighbor nature of , the only relevant expansion coefficients in Eq. (10) become those associated with the dimer bonding position, i.e., and . We further note that will in general be proportional to where is the dihedral angle between the two monomers. Finally, using intensity borrowing perturbation theory47, we find that the dipole moment of the CT excitation is, at first order,
[TABLE]
and the intensity of the new absorption will be proportional to the square of the dipole moment63.
4 Application to bipentacenes
Having arrived at an expression to estimate intensity borrowing to first order in Eq. (11), we undertake an investigation of the extent to which this phenomenon is observed in bipentacenes.
For 11BP a monomer calculation gives , such that and there is no new low-energy absorption. For 22BP a monomer calculation gives , and we find
[TABLE]
where we use the atom numbering in Fig. 1 and set eV and . Consequently,
[TABLE]
The perturbation therefore corresponds to of the UV peak intensity being borrowed. We emphasize that the 9% intensity borrowing calculated here is qualitative as we are neglecting all second-order contributions arising from mixing with other states. We further note that although the perturbation is sufficiently significant, leading to visible change in color, is still weak as evidenced by the UV absorption in 22BP and 22eBP continuing to be the dominant absorption. We further calculate the energy of the low-energy absorption from the first order correction to the energy of the CT state, which is eV, corresponding to the Coulomb attraction of an electron in the LUMO of one molecule and a hole in the HOMO of the other. This gives eV (464 nm), close to the experimentally observed new transition at 496 nm.
The dihedral angle arguments advanced earlier for the strength of the absorption also explain the more intense absorption seen in 22eBP. Treating the alkene linker to be a molecular ‘wire’ through which the monomers interact, we estimate the new absorption in 22eBP to be times greater than the absorption in 22BP. This is experimentally verified by the stronger visible absorption peak that appears in the spectra of Fig. 2A.
We find that the conclusions reached through intensity borrowing and perturbation theory (Eq. (11)) above can be anticipated by examining the nodal structure of the relevant monomer orbitals. In Fig. 3A, we present the top four HOMOs of TIPS-Pentacene obtained from an RHF calculation that are qualitatively similar to the orbitals from a PPP calculation. The HOMO–3 orbital has nodes on every long-axis carbon (red arrows in Fig. 3A), as is the case for all acenes8, and has been observed experimentally.74 The HOMO, HOMO–1 and HOMO–2 all have nodes in the horizontal () plane but the HOMO–3 does not. Now, consider joining together two monomers by the and positions, shown schematically in Fig. 3B. For 11BP in Fig. 3B(i), we see that the HOMO has nonzero amplitude at the position, but the HOMO–3 has zero amplitude such that the perturbation , leading to no new low-energy absorption peak in the spectrum. For 22BP, as shown in Fig. 3B(ii), both the HOMO and HOMO–3 have substantial amplitude, and a new low-energy absorption peak is predicted, and verified both by the experimental spectrum and from PPP calculations (Fig. 2).
Using similar arguments, once can show that indirect mixing (via another excitation) is likely to be minimal unless the monomers are joined via a short-axis carbon. The same inferences can be drawn by examining the monomer LUMOs using the Coulson-Rushbrooke theorem64, as shown in the SI.
The intensity borrowing and molecular orbital arguments above show that acene monomers must be joined via short-axis carbons in order to observe a new, low-energy absorption. Since this analysis can, in theory, be performed for arbitrarily many monomers, we would expect this result to hold for oligomers and polymers as well as dimers. In addition, the MO arguments suggest that joining acenes via a long-axis carbon or both short-axis carbons will not lead to enhanced low-energy absorption, since the HOMO has a long-axis () nodal plane whereas the HOMO–3 does not, leading to no constructive interaction between the relevant orbitals.
We are now ready to construct a simple design rule to make acene dimers, oligomers and polymers that will exhibit a new low-energy (visible) absorption peak in their spectra:
Join the monomers via a short-axis carbon, and avoid a long-axis symmetry plane passing through adjacent monomers.
This is the main result of the article and is summarized in Fig. 4.
Clearly, for the low-energy absorption to be significant there must be significant interactions between the monomers’ systems – they must be directly bonded, otherwise held very close together, or be connected via a conjugated linker (such as in 22eBP). Furthermore, if the design rule is satisfied, increasing planarity will increase the intensity of the extra absorption.
5 Application to general acenes
We have already demonstrated that the design rule proposed in the previous section can explain the presence or absence of a visible absorption peak in the spectra of 11BP, 22BP, and 22eBP. Here, we demonstrate the broad applicability of this rule by anticipating the experimental UV-Vis spectra of acene dimers, trimers, oligomers, and polymers, as well as a variety of hetero-atom substituted derivatives made for a broad range of applications including as a demonstration of organic synthesis 40, 33, 34, 44, for organic semiconductors 35, 36, 38, liquid crystals 41, polymer synthesis 42, sensors 45, photovoltaic applications 39, 37, 43, and one to explore structure-spectrum relationships 32.
A small selection of these are drawn in Fig. 5 grouped according to whether or not their spectra have enhanced low-energy absorption compared to their corresponding monomers. Our design rule correctly predicts the presence or absence of an extra absorption in all cases, including dimers, oligomers and polymers of naphthalene 32, anthracene (Fig. 5B and Fig. 5H 33, 40), tetracene (Fig. 5G 39) and pentacene (Fig. 1, Fig. 5A and Fig. 5F 37, 25, 59, 38), thereby explaining the structure-spectrum phenomenon first observed in 1948 32. It also correctly predicts the presence or absence of an extra absorption for unusual and complex geometries such as starphenes (Fig. 5C 34), iptycenes (Fig. 5I 41, 42), bridged dimers (Fig. 5J 43), and cyclophanes (Fig. 5K 44). Further, we find this design rule also holds in heteroatom-substituted acene derivatives such as the tetra-aza-pentacene dimer in Fig. 5D35, the anthrathiophene dimer in Fig. 5E36 and the aza-anthracene trimer Fig. 5L45 (compared to its corresponding monomers75, 76). This unexpected applicability of the design rule can be attributed to the ‘striped’ orbitals in Fig. 3A that will be solutions to the Hückel Hamiltonian of the heteroatom-substituted acene in cases where only the diagonal energies () are perturbed and not the off-diagonal () terms. In general, this will hold when the the heterosubstitution is only on long-axis carbons. For the thiophene derivative, standard organic chemistry77 suggests that the S heteroatom is of a similar size to a C=C double bond, making thiophene qualitatively similar to benzene. With this reasoning the anthrathiophene dimer can be likened to a -tetracene dimer, explaining the new low-energy absorption.
Finally, we note that it is challenging to apply the design rule described here to oligomers where two or more moieties overlap in such a way that through-space interaction become important, such as the cross-conjugated dimers in Refs 26 and 78. In these cases a more sophisticated formulation of the hopping term than nearest-neighbor interactions will be required for PPP theory (and the intensity borrowing arguments) to be applied.
6 Conclusions
This article uses PPP theory and intensity borrowing perturbation theory to construct a simple rule for predicting and explaining the low-energy absorption spectra of acene derivatives. The resulting testable and experimentally verifiable design rule has been found to hold in a large variety of dimers, trimers, oliogmers and polymers, including those with heteroatom substitution and unusual bonding geometries. This is particularly useful because it allows for the presence or absence of a new low-energy absorption to be determined from the monomer orbitals alone, without having to simulate the electronic structure of each dimer or oligomer separately. This a priori design of highly absorbent molecules, of which this is a proof-of-concept, has significant implications for photovoltaic design, where some organic solar photovoltaics are constrained by the small diffusion length of excitons compared to the thickness of material required to absorb a significant fraction of visible light22. Developing more absorbent molecules that retain the necessary photophysical charge-transport properties could allow for thinner, flexible, cheaper and more efficient solar cells.
{acknowledgement}
We thank Roald Hoffmann and Tao Zeng for helpful discussions and acknowledge spectra from Dan Lehnherr and Rik Tykwinski. We also thank Robert A. DiStasio Jr. and Stuart Althorpe for helpful comments on the manuscript. We thank the Nuckolls lab for use of their UV-vis spectrophotometer. NA acknowledges funding from NSF CAREER grant (Award No. CHE-1555205) and a Sloan Foundation Research Fellowship. TJHH acknowledges funding from NSF EAGER grant (Award No. CHE-1546607) and from Jesus College, Cambridge. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), supported by National Science Foundation grant number ACI-1053575. L.M.C. acknowledges support from the Office of Naval Research Young Investigator Program (Award N00014-15-1-2532), ACS Petroleum Research Fund, 3M Non-Tenured Faculty Award, Arthur C. Cope Scholar Award, and Cottrell Scholar Award. SNS thanks the NSF for GRFP (DGE 11-44155). This research used resources of the Center for Functional Nanomaterials, which is a U.S. DOE Office of Science Facility, at Brookhaven National Laboratory under Contract No. DE-SC0012704.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hückel 1930 Hückel, E. Zur Quantentheorie der Doppelbindung. Zeitschrift für Physik 1930 , 60 , 423–456
- 2Thomas 1925 Thomas, W. Über die Zahl der Dispersionselektronen, die einem stationären Zustande zugeordnet sind. (Vorläufige Mitteilung). Naturwissenschaften 1925 , 13 , 627–627
- 3Reiche and Thomas 1925 Reiche, F.; Thomas, W. Über die Zahl der Dispersionselektronen, die einem stationären Zustand zugeordnet sind. Zeitschrift für Physik 1925 , 34 , 510–525
- 4Kuhn 1925 Kuhn, W. Über die Gesamtstärke der von einem Zustande ausgehenden Absorptionslinien. Zeitschrift für Physik 1925 , 33 , 408–412
- 5Pople 1953 Pople, J. A. Electron interaction in unsaturated hydrocarbons. Trans. Faraday Soc. 1953 , 49 , 1375–1385
- 6Pople 1955 Pople, J. A. The Electronic Spectra of Aromatic Molecules II: A Theoretical Treatment of Excited States of Alternant Hydrocarbon Molecules based on Self-Consistent Molecular Orbitals. P. Phys. Soc. Lond.. Section A 1955 , 68 , 81
- 7Pariser and Parr 1953 Pariser, R.; Parr, R. G. A Semi-Empirical Theory of the Electronic Spectra and Electronic Structure of Complex Unsaturated Molecules. I. J. Chem. Phys. 1953 , 21 , 466–471
- 8Pariser 1956 Pariser, R. Theory of the Electronic Spectra and Structure of the Polyacenes and of Alternant Hydrocarbons. J. Chem. Phys. 1956 , 24 , 250–268