Quantum gas microscopy of Rydberg macrodimers
Simon Hollerith, Johannes Zeiher, Jun Rui, Antonio Rubio-Abadal,, Valentin Walther, Thomas Pohl, Dan M. Stamper-Kurn, Immanuel Bloch, Christian, Gross

TL;DR
This paper demonstrates the direct microscopic observation and control of Rydberg macrodimers in an ultracold atomic gas, enabling detailed study of their properties and interactions at unprecedented spatial resolution.
Contribution
It introduces the use of quantum gas microscopy to directly image and characterize Rydberg macrodimers, revealing over 50 vibrational resonances and enabling control of molecular alignment.
Findings
Resolved more than 50 vibrational resonances.
Observed macrodimers via correlated atom loss.
Controlled molecular alignment through vibrational state selection.
Abstract
A microscopic understanding of molecules is essential for many fields of natural sciences but their tiny size hinders direct optical access to their constituents. Rydberg macrodimers - bound states of two highly-excited Rydberg atoms - feature bond lengths easily exceeding optical wavelengths. Here we report on the direct microscopic observation and detailed characterization of such macrodimers in a gas of ultracold atoms in an optical lattice. The size of about 0.7 micrometers, comparable to the size of small bacteria, matches the diagonal distance of the lattice. By exciting pairs in the initial two-dimensional atom array, we resolve more than 50 vibrational resonances. Using our spatially resolved detection, we observe the macrodimers by correlated atom loss and demonstrate control of the molecular alignment by the choice of the vibrational state. Our results allow for precision…
Click any figure to enlarge with its caption.
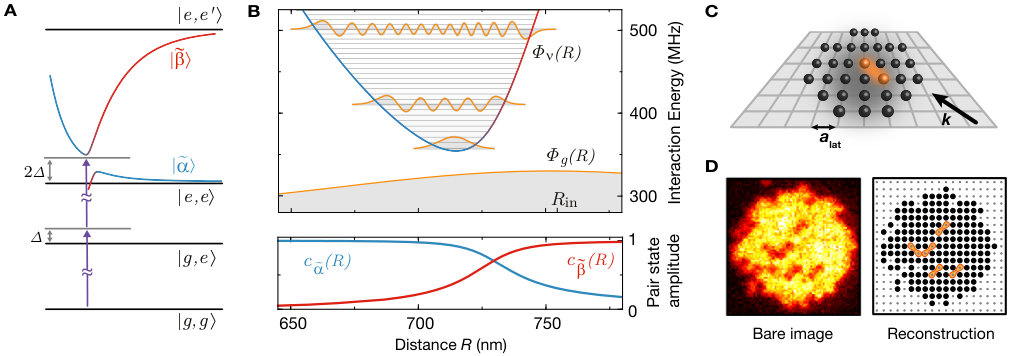 Figure 1
Figure 1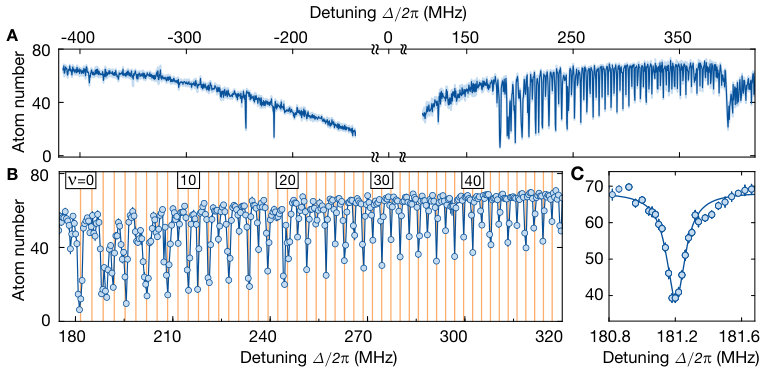 Figure 2
Figure 2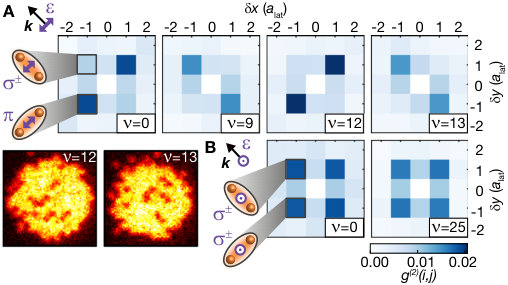 Figure 3
Figure 3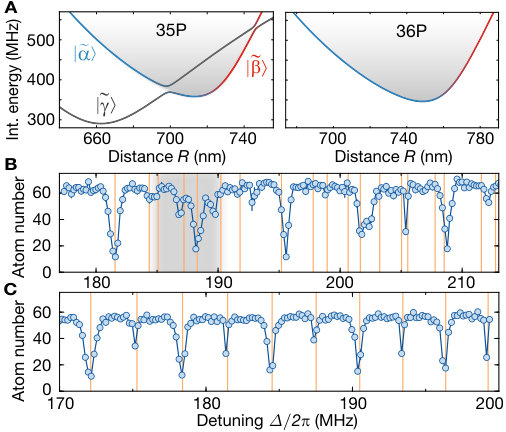 Figure 4
Figure 4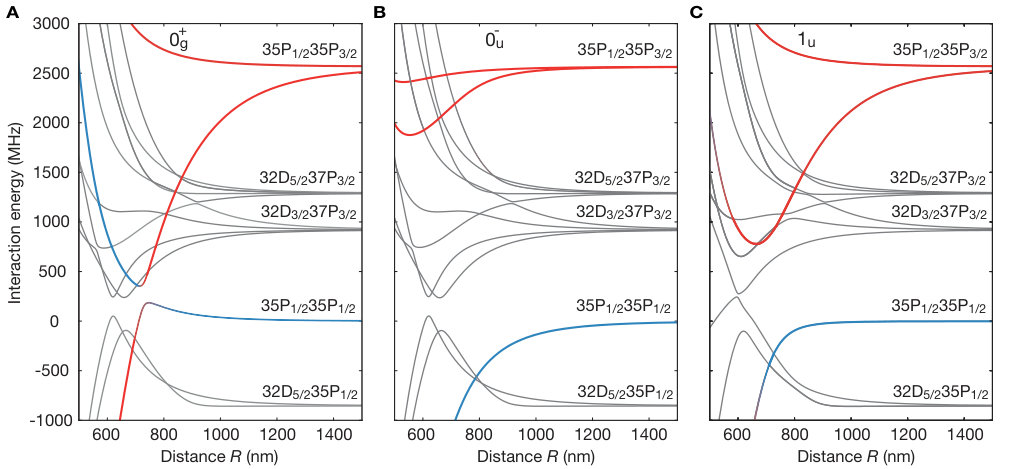 Figure 1
Figure 1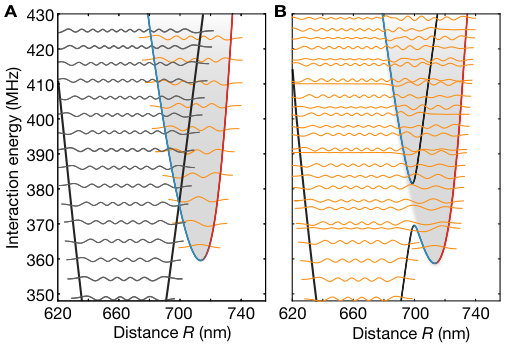 Figure 2
Figure 2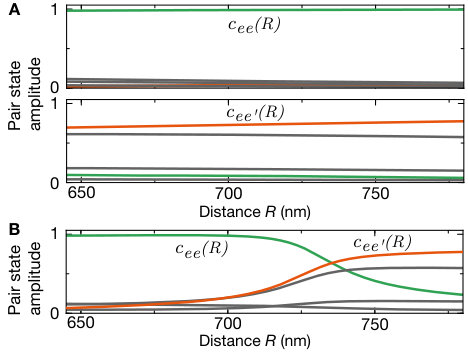 Figure 3
Figure 3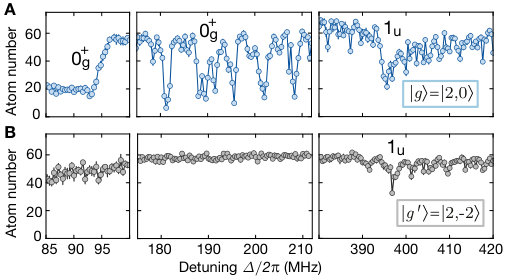 Figure 4
Figure 4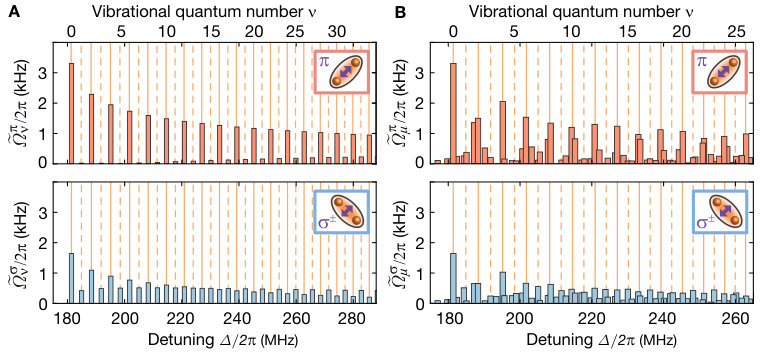 Figure 5
Figure 5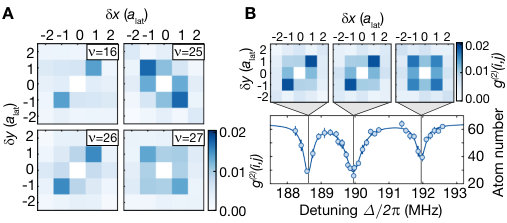 Figure 6
Figure 6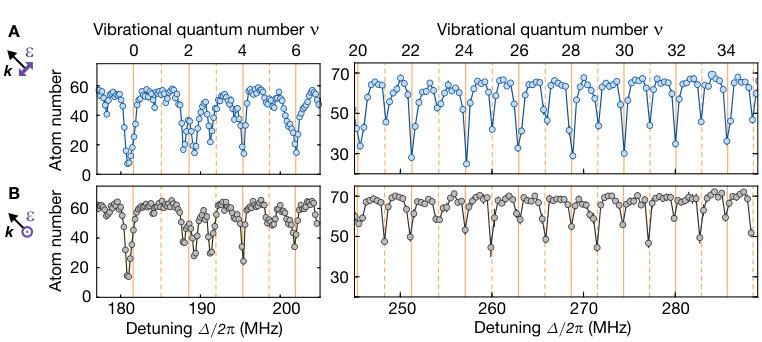 Figure 7
Figure 7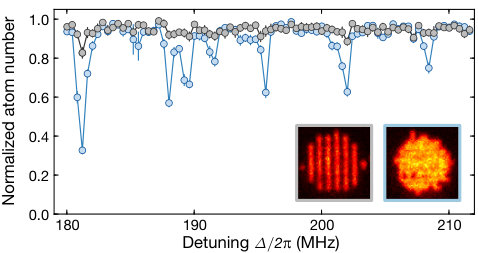 Figure 8
Figure 8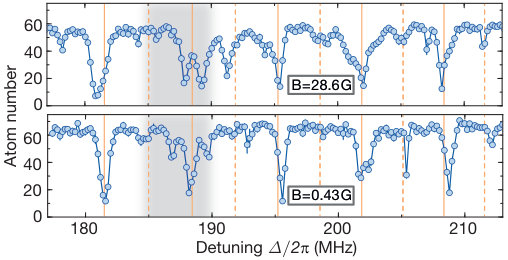 Figure 9
Figure 9| even | + | + | - | strong | weak |
| odd | - | + | - | weak | strong |
| 0 | 9 | 12 | 13 | 16 | 25 | 26 | 27 | |
|---|---|---|---|---|---|---|---|---|
| 0.12 | 5.0 | 1.5 | 10 | 0.9 | 10 | 3.5 | 3 | |
| Exp. shots | 158 | 240 | 571 | 283 | 197 | 274 | 256 | 264 |
| Filling | 87.7(5) | 88.4(3) | 87.6(3) | 87.4(5) | 89.7(4) | 85.1(4) | 86.5(5) | 86.6(4) |
| 1.90(17) | 0.64(10) | 2.05(08) | 0.49(10) | 1.39(12) | 0.61(11) | 1.53(11) | 1.01(10) | |
| 0.95(14) | 1.51(13) | 0.72(06) | 1.45(11) | 0.59(11) | 1.74(13) | 0.52(10) | 1.35(12) |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCold Atom Physics and Bose-Einstein Condensates · Spectroscopy and Laser Applications
\sidecaptionvpos
figureh
††thanks: present address: Department of Physics, University of California, Berkeley, California 94720, USA
Quantum gas microscopy of Rydberg macrodimers
Simon Hollerith
Max-Planck-Institut für Quantenoptik, 85748 Garching, Germany
Johannes Zeiher
Jun Rui
Max-Planck-Institut für Quantenoptik, 85748 Garching, Germany
Antonio Rubio-Abadal
Max-Planck-Institut für Quantenoptik, 85748 Garching, Germany
Valentin Walther
Department of Physics and Astronomy, Aarhus University, DK 8000 Aarhus C, Denmark
Thomas Pohl
Department of Physics and Astronomy, Aarhus University, DK 8000 Aarhus C, Denmark
Dan M. Stamper-Kurn
Department of Physics, University of California, Berkeley, CA 94720, USA
Immanuel Bloch
Max-Planck-Institut für Quantenoptik, 85748 Garching, Germany
Fakultät für Physik, Ludwig-Maximilians-Universität München, 80799 München, Germany
Christian Gross
Max-Planck-Institut für Quantenoptik, 85748 Garching, Germany
(2nd March 2024)
Abstract
The sub-nanoscale size of typical diatomic molecules hinders direct optical access to their constituents. Rydberg macrodimers — bound states of two highly-excited Rydberg atoms — feature interatomic distances easily exceeding optical wavelengths. Here we report the direct microscopic observation and detailed characterization of such molecules in a gas of ultracold atoms in an optical lattice. The bond length of about , comparable to the size of small bacteria, matches the diagonal distance of the lattice. By exciting pairs in the initial two-dimensional atom array, we resolve more than 50 vibrational resonances. Using our spatially resolved detection, we observe the macrodimers by correlated atom loss and demonstrate control of the molecular alignment by the choice of the vibrational state. Our results allow for rigorous testing of Rydberg interaction potentials and highlight the potential of quantum gas microscopy for molecular physics.
A quantitative determination of the structure of molecules is an essential goal of physical chemistry and is crucial to reveal and understand their properties. The high level of quantum control and the ultracold temperatures achieved in atomic physics provide novel tools to study molecules and their structure Jones et al. (2006). Prominent examples include the observations of weakly bound Feshbach molecules Regal et al. (2003); Moses et al. (2015), the controlled photoassociation of individual molecules in a microtrap Liu et al. (2018), molecules comprising ground-state atoms bound to a highly excited Rydberg atom Gallagher (1994); Bendkowsky et al. (2009); Shaffer et al. (2018) or pure long-range molecules Miller et al. (1993); Lett et al. (1993) which are bound purely electrostatically. The binding mechanism of the latter, where the electron orbitals of the constituents do not overlap, has also been predicted to exist between two Rydberg atoms. These “Rydberg macrodimers” Boisseau et al. (2002); Stanojevic et al. (2006, 2008); Schwettmann et al. (2006, 2007) are truly remarkable in their macroscopic bond lengths, 10 000 times larger than usual diatomic molecules and thus reaching typical interparticle distances in magneto-optical traps Saßmannshausen and Deiglmayr (2016), optical lattices Guardado-Sanchez et al. (2018) or tweezers Bernien et al. (2017); Labuhn et al. (2016). Their enormous size not only enables direct optical access to individual constituents, but also allows for the controlled binding of two atoms optically pinned at the correct distance. First signatures of Rydberg macrodimers have been observed in systems of laser-cooled atoms by spectroscopy Saßmannshausen and Deiglmayr (2016) and pulsed-field ionization Overstreet et al. (2009), but a vibrationally and spatially resolved detection has been lacking so far.
Here, we present a precise study of Rydberg macrodimers starting from ground-state atoms deterministically arranged in an optical lattice. We probe the vibrational levels by two-photon spectroscopy, resolving more than 50 excited vibrational resonances. The observed spectrum agrees quantitatively with ab initio calculations of Rydberg interaction potentials Weber et al. (2017); S̆ibalić et al. (2017); Deiglmayr (2016), providing a stringent test for their accuracy. Using the site-resolved detection and single-atom sensitivity of our quantum gas microscope Sherson et al. (2010); Bakr et al. (2010), we identify the macrodimer signal microscopically as a loss of pairs of ground state atoms at a distance of a bond length. Furthermore, we control the spatial orientation of the photo-associated molecules by the parity of the vibrational wave function and the polarization of the excitation laser.
While Rydberg interaction potentials are of van-der-Waals type for asymptotically large interatomic separations Saffman et al. (2010), the situation is more complicated at smaller distances. In a generic situation, repulsive interactions may increase the energy of a lower lying pair state , while attractive interactions decrease the energy of a higher lying one . At some specific distance the two potentials become degenerate and any finite coupling between them opens a gap (see Fig. 1 A), which is large when and contain significant amplitudes of mutually dipole-dipole coupled states. The resulting potential minimum hosts a series of bound macrodimer states Boisseau et al. (2002); Stanojevic et al. (2006); Saßmannshausen and Deiglmayr (2016), where denotes the vibrational quantum number of the nuclear motion. In our experiment we choose such an avoided crossing of two potentials with gerade symmetry and zero angular momentum projection on the interatomic axis Jones et al. (2006); Weber et al. (2017). For large distances, the selected pair states transform into the states and SI , which can be optically coupled from the ground state by a two photon transition. Here, and the bond length of this macrodimer state is predicted to be nm, close to the diagonal lattice spacing of nm in our optical lattice. With the atoms being initially prepared in the motional ground state of the optical lattice, this coincidence of length scales results in a strong optical coupling due to the large wave function overlap.
Our experiments started with a two-dimensional atomic Mott insulator of with a lattice filling of in the atomic ground state. The atoms were pinned in a deep optical lattice with a rms width nm of the motional ground state in the atomic plane (see Fig. 1 B) with a temperature below the on-site trapping frequency. The molecules were photoassociated by an ultraviolet (UV) Rydberg excitation laser at a wavelength of nm propagating along the diagonal direction of the optical lattice, with linear polarization aligned either in or orthogonal to the lattice plane. Our typical optical Rabi coupling from the state to was MHz. We detected the excited macrodimers as missing pairs of ground state atoms as these are ejected from the optical lattice very efficiently due to the release of kinetic energy in the macrodimer decay. The remaining ground state atoms were then imaged with near-unity fidelity using a quantum gas microscope, see Fig. 1 D.
We first aimed at identifying the presence of the bound macrodimers by their spectroscopic fingerprint. To this end, we illuminated the atomic ensemble for ms with the UV light polarized in the atomic plane for varying detuning from the bare Rydberg state . In each step, we swept the optical frequency of the excitation laser by kHz during the illumination time to ensure coverage of the full spectral region between neighboring data points. We observe the coupling to the bare Rydberg state as a very broad saturated loss resonance, see Fig. 2. At negative detunings, the resonance features an asymmetric broadening due to coupling to attractively interacting pair-state potentials Singer et al. (2004). At positive detunings around MHz, the first macrodimer bound state becomes two-photon resonant. A non-saturated high-resolution spectroscopy of this line (see Fig. 2 C) yields a FWHM of kHz, which is of the same order as the measured linewidth of the bare Rydberg resonance Zeiher et al. (2016). For higher vibrational resonances we observed a reduction of the line strength, which we attribute to a combination of increased intermediate-state detuning and reduced overlap of the spatial wave functions. In addition, we observed a suppression of the excitation to odd vibrational states due to the approximate odd parity symmetry of the molecular wave functions with respect to the equilibrium distance. This is consistent with the Franck-Condon principle, which predicts the coupling to be proportional to the overlap integrals of the broad initial and the tightly confined final spatial wave functions and , see Fig. 1 B. However, a closer inspection of the experimental data shows that this simple picture needs to be refined. Repeating the spectroscopy with orthogonal polarization (so the optical electric field oscillates out of the atomic plane) results in a suppression of the line strength of the even lines.
Our microscopic access provides direct in situ information about the spatial alignment of the associated molecules and valuable insights into the underlying coupling mechanism Samboy and Côté (2011); Samboy et al. (2011); SI . We compared different molecular lines by illuminating the cloud with UV light, resonant with a given vibrational state , until the filling of the lattice decreased to roughly . For a quantitative analysis, we evaluated spatially averaged density-density correlations for the measured spatial atom distributions on a region of interest of lattice sites, see Fig. 3. Correlations show a clear peak at a distance of a lattice diagonal, revealing the bond length of the molecule. Moreover, we controlled the orientation of the photo-associated molecules by choosing a combination of vibrational quantum number and polarization of the light field. For even oscillator states , the correlations are stronger along the lattice diagonal parallel to the polarization of the excitation light. The molecular orientation, however, flips when considering odd oscillator states for which the dimers form predominantly along the direction perpendicular to the polarization.
The key to understanding this striking alternation in the orientation of the molecules is the interplay of electronic and motional degrees of freedom. The Born-Oppenheimer wave function of the macrodimer can be expressed as . Whereas at large distances, the electronic part Stanojevic et al. (2006, 2008) is dominated by the state , for short distances the contribution dominates (see Fig. 1 B), and this parametric dependence of the electronic potential has to be taken into account in the optical excitation. The two-photon Rabi coupling from the ground state to the macrodimer states thus splits into two terms. Neglecting the weak spatial dependence of the two-photon Rabi couplings to the states , we obtain with the generalized Franck-Condon integrals and Samboy and Côté (2011); SI . Because of the change of the pair state amplitudes around the potential minimum (see Fig. 1 B), and have the same sign for even but an opposite sign for odd . The electronic contributions depend on the alignment of the polarization relative to the molecular axis because the states obey molecular symmetry contraints. In the case where the axes are parallel, and have the same sign and therefore both terms in add constructively for even vibrational states, leading to a dominating signal in . For the perpendicular case, the sign of flips and constructive interference occurs for odd vibrational states, resulting in a stronger value for g^{(2)}(1,\scalebox{0.75}[1.0]{-}1). Moreover, for the measurement with light polarized out of plane (see Fig. 3 A), the spatial signal exhibits isotropic correlations because in that case the polarization is perpendicular to both lattice diagonals.
Although most of the line positions of the measured spectrum shown in Fig. 2 agree with the theoretical model, we find deviations for low-lying oscillator states. A finer scan of that region is shown in Fig. 4 A. These deviations originate from a third pair state asymptotically corresponding to the optically uncoupled state intersecting the binding potential around the potential minimum. At the degeneracy point, the weak dipole-quadrupole coupling between the intersecting potentials opens another gap energetically comparable to the vibrational energy. As a consequence, a separation of the vibrational motion and interatomic interaction is no longer possible. For the theory used to describe the coarse vibrational structure shown in Fig. 2, we only accounted for the crossing formed by the coupling between and . We extended our theory by allowing for the vibronic coupling between the vibrational modes and the electronic states , and . The modified eigenenergies are indicated as orange lines in Fig. 4 B. Using the refined theory, we indeed can identify almost all observed lines. To confirm the effect of the intersection, we repeated our spectroscopic measurements for the lowest states in the analogous potential for , where such an additional crossing is absent. In this case, we observed a pure harmonic-oscillator-like spectrum in excellent agreement with the calculations. The remarkably high sensitivity of the measured line structure to even weak modifications of the interaction potentials underlines the promise of Rydberg macrodimer spectroscopy for benchmarking Rydberg interaction potentials. This also holds for Rydberg interactions in the presence of applied magnetic fields, where accurate calculations are more difficult SI .
In the future, the coupling to macrodimers could be used to realize quantum gates at well-defined qubit distance or to enhance Rydberg dressing schemes van Bijnen and Pohl (2015); Zeiher et al. (2016), where the pair state admixture of the strongly interacting doubly excited state is significantly enhanced compared to the singly-excited intermediate states. The strongly spatially dependent loss revealed in the correlation measurements and also by modulating the initial atom distribution (SI, ) could be used to engineer dissipatively stabilized few- or many-body states Ates et al. (2012); Syassen et al. (2008). Furthermore, the approach demonstrated here can readily be extended to study multi-atom bound states Samboy and Côté (2013); Kiffner et al. (2013), also in optical tweezers. Finally, bringing the coupling rate to the macrodimers closer to the decay rate of the individual Rydberg atoms may allow for the observation of novel many-body physics arising from spatial constraints and coherent interactions.
Acknowledgements.
Acknowledgements: We thank all contributors to the open-source programs “pair interaction” and “ARC” as well as Robin Côté, Bill Phillips, Nikola Šibalić and Johannes Deiglmayr for valuable discussions. We acknowledge support by the DNRF through a Niels Bohr Professorship for T.P. and funding by MPG. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 817482 (PASQuanS) and the European Research Council (ERC) No. 678580 (RyD-QMB) and also from the project No. 319278 (UQUAM) of the Seventh Framework Programme. We also acknowledge funding from Deutsche Forschungsgemeinschaft (Project No. BL 574/15-1) within SPP 1929 (GiRyd).
Author contributions: All authors contributed significantly to the work presented in this manuscript.
Data availability: The data that support the plots presented in this paper are publicly available from the Open Access Data Repository of the Max Planck Society (https://edmond.mpdl.mpg.de) dat
Competing interests: The authors declare no competing interests.
Supplementary Materials
This Supplementary Information contains the calculation of the expected positions of the macrodimer resonances, the wave functions and the optical coupling, additional measurements supporting the statements in the main text and experimental details.
I Calculation of the macrodimer spectrum
From the discussion of Fig. 2 and Fig. 4 B, it becomes clear that a calculation in the two isolated potential curves agrees well with the measured spectrum of the macrodimers at high vibrational quantum numbers. However, for low-lying vibrational states, where the vibrational energy is comparable to the dipole-quadrupole coupling between the intersecting potentials, the vibronic coupling between them becomes relevant. This leads to a breakdown of the Born-Oppenheimer approximation and we expect deviations from a description in the isolated potentials. For convenience, we call the uncoupled potentials diabatic and the coupled potentials adiabatic. In the following, we describe the calculation of the uncoupled, diabatic electronic eigenstates relevant for higher vibrational states from coupled, adiabatic states obtained from the diagonalization of the Rydberg interaction Hamiltonian (Weber et al., 2017). In contrast to the discussion in the main text, we cannot restrict our discussion to a single potential curve, leading to additional indices and (see below). Subsequently, we present the calculation of the exact vibronic wave functions by recoupling the obtained purely electronic diabatic eigenstates and purely vibrational states calculated in these diabatic potential wells.
The total atomic Hamiltonian
[TABLE]
can be split into an electronic part describing the spectrum of each of the atoms ( and ) as well as the electrostatic interactions between them, and a term that represents the relative kinetic energy of the atom pair. At interatomic distances larger than the LeRoy radius Roy (1974), the electrostatic interaction in the electronic part can be calculated by diagonalizing the interaction Hamiltonian obtained from a multipole expansion at various interatomic distances . The results are adiabatic electronic Born-Oppenheimer potentials , given by
[TABLE]
In position representation, the electronic eigenstates are written as where acts as a parameter. The eigenstates can be expanded in asymptotic pair states , which are the eigenstates of the interaction in the asymptotic limit of infinite interatomic distance,
[TABLE]
The resulting adiabatic potential curves obtained for the macrodimer potentials relevant for this work are plotted in Fig. S1. In the following we calculate the diabatic states used in the discussion in the first part of the main text. The coupling of the adiabatic eigenstates is provided by the kinetic energy operator , where is the atomic mass. Using the adiabatic states as a basis, we can expand the total wave function
[TABLE]
where are distance-dependent amplitudes of adiabatic states which take into acount the nuclear motion. Substitution into the Schrödinger equation of the total system then yields the matrix equation Domcke et al. (2004)
[TABLE]
where the non-adiabatic couplings are defined by
[TABLE]
For isolated interaction potentials where the non-adiabatic couplings are typically negligible (as for in Fig. 4 C), Eq. (S5) can be solved directly for the vibronic eigenstates and corresponding energies. In this case, the amplitudes can be interpreted as pure vibrational states in the adiabatic potential wells. In our case, the non-adiabatic couplings are significant. In order to use a shorter notation we define and the coupling matrices and with respective elements and . Further, we introduce and use the notation and . We can write Eq. (S5) as
[TABLE]
where we use the identity to re-express . With this form of the Schrödinger equation, the matrix can be interpreted as a gauge potential. Indeed we can apply a unitary transformation and obtain the same equation
[TABLE]
with
[TABLE]
We can now use this transformation to eliminate the additional derivative terms by choosing such that
[TABLE]
In the description of the diabatic basis obtained by such a transformation, the coupling by the kinetic energy of the nuclei vanishes but becomes off-diagonal Pacher et al. (2007). A calculation in a purely diabatic picture can be done by neglecting the obtained off-diagonal terms.
For explicit calculations we can isolate two adjacent adiabatic potential curves, labeled here as with . In this basis we have
[TABLE]
and can parametrize our unitary transformation as
[TABLE]
Substituting this into our Eq. (S10) yields
[TABLE]
The gauge transformation fulfilling Eq. (S13) is thus given by
[TABLE]
where is chosen to be far outside of the coupled region. The two new quasi-diabatic basis states are given by
[TABLE]
via Eq. (S12). In this basis, we then obtain a two-component Schrödinger equation
[TABLE]
with
[TABLE]
Eq. (S16) can be solved conveniently in two steps. We first solve the Schrödinger equation for each diagonal potential and numerically to obtain the set of vibrational wavefunctions at energies related with the diabatic potential (see Fig. S2 A for the potential relevant in this work), leading to the molecular states
[TABLE]
in both diabatic potentials. Here, the nuclear motion is treated independently from the electronic interaction petential in a Born-Oppenheimer framework. Now, the residual coupling due to the off-diagonal terms is then accounted for by diagonalizing the matrix in the basis of the , which gives the vibronically coupled eigenstates (see Fig. S2 B)
[TABLE]
Inserting Eq. (S15) into Eq. (S19), the vibronically coupled eigenstates can then be re-expressed as
[TABLE]
with . In the following calculations of the optical coupling to the macrodimers in our potential, we omit the index in Eq. (S18) because a diabatic description is only relevant for the optically coupled eigenstates in the potential , see Fig. S2 A.
II Optical coupling
In the main text, we described the macrodimer potential by the three crossing van-der-Waals states , and . The weak dependence on the interatomic distance due to dispersive van-der-Waals interactions between off-resonant asymptotic pair states was neglected in the main text. Due to this weak dependence, the two-photon Rabi couplings are sligthly spatial dependent because of the changing state admixture in . Here, we use the expansions Eq. (S18) and Eq. (S20) into asymptotic pair states instead. These asymptotic pair states are the eigenstates in the non-interacting limit of large separations. Since it is straightforward to expand the asymptotic states into products of single-particle states, optical coupling rates to these pair states are easy to calculate. As we will see in Eq. (S27), this allows for a strict separation of spatially independent optical couplings to contributing pair states and generalized Franck-Condon overlap integrals taking into account the spatial dependent amplitudes of these pair states.
The two dominating asymptotic pair states contributing to our potential are the non-interacting pair states and with coefficients and , which are the asymptotic limits of the van-der-Waals states and . In the main text, these asymptotic states were defined via the states and . Accounting also for the spin projections of both atoms and symmetrizing the states with respect to the inversion and reflection symmetry of the (Weber et al., 2017) potential leads to
[TABLE]
[TABLE]
with spin orientations and relative to the molecular axis. A distance dependent decomposition of the states and which form the potential well is shown in Fig. S3. Among other states, we find an optically uncoupled pair state including single particle states and and weaker admixed pair states, where both atoms are in . The optical coupling to the latter are suppressed not only by smaller pair state amplitudes, but also because they are only accessible via the intermediate state . Due to the fine-structure splitting of MHz for our Rydberg state, ten times larger compared to the typical detunings to , we restrict ourself to the intermediate pair states states including one atom in the ground state and a second atom in or in the following calculations.
II.1 Molecular symmetry
This work mainly discusses the macrodimers in the potential with gerade symmetry and zero angular momentum projection on the intermolecular axis. Interestingly, the antisymmetric states (see definition of and in Eq. (S21) and Eq. (S22)) in the potential can only be optically coupled because of the hyperfine coupling of the ground state, which is negligible for Rydberg states. This is due to the fact that the initial pair states are symmetric and that the optical coupling preserves the pair state symmetry. Rewriting the initial pair state with into the components of the electronic angular momentum and the nuclear spin leads to
[TABLE]
Here, only the antisymmetric part \lvert\scalebox{0.75}[1.0]{-}\rangle_{J}=1/\sqrt{2}(\lvert 5S_{1/2}\uparrow,5S_{1/2}\downarrow\rangle-\lvert 5S_{1/2}\downarrow,5S_{1/2}\uparrow\rangle) can couple to the desired potential. We probe this experimentally by starting with the initial pair state with which does not contain antisymmetric fine-structure components, as shown in Fig. S4. All other experimental parameters are identical to the spectroscopy with initial state . As expected, we find spectroscopic features for the lower and the upper branch of the avoided crossing in the potential only by starting from , while the symmetric potential can be observed for both initial states. Eq. (S23) also implies that our photoassociation procedure projects both nuclear spins into the entangled state .
II.2 Calculation of Rabi frequencies
The Hamiltonian describing the optical coupling is given by
[TABLE]
where and are the position operators of both electrons and is the electric field of the excitation light. For the calculation of the optical couplings to the macrodimers (Samboy and Côté, 2011; Samboy et al., 2011) we have to account for the fact that the eigenstates of the interatomic interaction are only symmetric with respect to rotations around the molecular axis but not necessarily spherical symmetric. Therefore, the optical couplings to pair states depend on the relative orientation of the polarization of the light and the molecular axis. Hence, we have to rotate our initial state , which is defined with respect to the initial quantization axis of the magnetic field to be parallel to the quantization axis of the molecule where the states are defined. Here, we make use of the decomposition (S23) into optically uncoupled symmetric and coupled antisymmetric initial pair states where \lvert\scalebox{0.75}[1.0]{-}\rangle_{J} is formally equivalent to a rotationally symmetric spin singlet state. This allows us to use \lvert\scalebox{0.75}[1.0]{-}\rangle_{J} as the initial state for all molecular orientations and the only parameter which depends on the interatomic axis is the polarization of the excitation light.
In the case of a polarization parallel to the molecular axis, the light is -polarized and the only relevant intermediate states which can couple to the potential (see Eq. (S21) and Eq. (S22)) are antisymmetric states and . The coupling strength from to the intermediate state is given by
[TABLE]
where we used that the coupling elements to both intermediate states turns out to be the same. For the coupling from the intermediate states to the macrodimer states, we have to take into account the different pair states contributing to Eq. (S18). Additionally, generalized Franck-Condon integrals now appear, with the initial relative wave function of two nuclei in the lattice Bloch et al. (2008)
[TABLE]
Here, is the extension of the on-site ground state wave function for a single atom and is the diagonal distance in the lattice. This leads to
[TABLE]
where we introduced the spatially independent optical couplings to the contributing non-interacting pair states . Applying a rotating-wave approximation in a frame co-rotating with the laser frequency, the full coupling Hamiltonian, in the basis , reads
[TABLE]
if the laser is two-photon resonant (i.e. ) to a macrodimer state . Since , the two intermediate states can be adiabatically eliminated, leading to a total effective Rabi coupling from to
[TABLE]
Within a simplified description where we only account for the dominating asymptotic states and , the sum reduces to
[TABLE]
with the experimentally calibrated single-photon coupling rate MHz and a numerical factor taking into account slightly different reduced matrix elements between the ground state and the two fine-structure states and .
In the case of a polarization perpendicular to the molecular axis, the light is -polarized and the intermediate states used above cannot be coupled. Now, we find and . An analogous theoretical analysis leads to
[TABLE]
which again can be approximated by
[TABLE]
Comparing both approximated equations, one can recognize a flip in the relative sign between the optical couplings to and , which is the origin of the alternating molecular orientation, see Table S1. It turns out that Eq. (S30) and Eq. (S32) capture all essential physics to understand the optical coupling mechanism and only slightly deviates from a more accurate calculation.
This is not only because of the strong domination of the states and in the pair potentials but also because other contributing pair states experience absent or strongly supressed optical couplings.
A rigorous calculation using Eq. (S29) and Eq. (S31) and taking into account a large set of basis states as well as the experimentally calibrated Rabi frequency MHz is shown in Fig. S5 A. While the discussion so far was focused on states in the diabatic potential , the optical couplings and to the vibronically coupled states can be calculated by replacing the pair state amplitudes and vibrational wave functions in the generalized Franck-Condon integrals by the coefficients defined in Eq. (S20). The result of this calculation is shown in Fig. S5 B. Besides a splitting of the eigenergies in the diabatic potential due to the vibronic coupling, the coarse structure and the polarization dependence remain qualitatively the same.
II.3 Further experimental tests
In Fig. S6 A, we show additional correlation measurements with the same behaviour as presented in the main text for higher vibrational quantum numbers up to . Interestingly, the concept of even and odd vibrational wave functions breaks down around the intersection (see Fig. S6 B) where the splitting of the lines due to the vibronic coupling does not allow a classification in alternating even and odd vibrational quantum numbers. Measured correlations at the three resonances around MHz show that the two left resonances feature almost the same directionality in the correlations, while the right resonance at MHz features almost isotropic correlations.
As briefly mentioned in the main text, rotating the UV polarization out of plane modifies the relative strength of even and odd macrodimer resonances, see Fig. S7. This can be understood from the microscopic picture developed above. For the configuration shown in Fig. S7 A, molecule formation occurs due to coupling rates as well as . Because of the strong coupling for even vibrational states, these states dominate the total loss spectrum, see Fig. S5. Additionally, as predicted and verified in the correlation measurements, losses at even(odd) resonances occur mainly due to molecule formation parallel(perpendicular) to the polarization of the UV light. In the case of Fig. S7 B, both molecular orientations are orthogonal to the dimer axis and we only probe . We find that even resonances are weaker but still dominating for low vibrational states. However, consistent with the calculations shown in the lower panels of Fig. S5, this flips for higher vibrational states where the odd resonances become stronger.
Furthermore, we probed the strong distance dependence of the losses by changing the spatial configuration of the atoms in the lattice. We use our ability to locally remove atoms from our initial Mott insulator by optically adressing individual lattice sites with our microscope Weitenberg et al. (2011) to create an initial pattern of alternatingly populated and empty rows in the optical lattice for which atom pairs at a distance of are absent. For both initial configurations, we probe the first macrodimer resonances spectroscopically, see Fig. S8. As expected, the formation of macrodimers is strongly suppressed in the new density modulated configuration. A closer look reveals that the atom loss at the macrodimer resonances is still larger compared to the background loss due to the off-resonant coupling to the bare Rydberg resonance. Measuring decay constants for both cases shows that we can suppress the decay by roughly one order of magnitude by density modulating the initial Mott insulator. The remaining coupling is consistent with the macrodimer excitation of two atoms at neighboring lattice sites, where the Franck-Condon factor is much smaller but not zero.
III Experimental details
For most of the data presented in this work (also for ), we applied a magnetic field G perpendicular to the atomic plane. At this field strength, the Rydberg state splits into two Zeeman sublevels separated by MHz. The magnetic field is expected to be not a critical parameter for our potential with zero angular momentum projection, even if the intermediate states get Zeeman splitted. This was verified by comparing the spectroscopic signal at low vibrational quantum numbers for two different field values, see Fig. S9. As expected, the energy of the diabatic eigenstates stays mainly unaffected. However, we find that the splitting of the lines with respect to the diabatic eigenergies due the intersecting potential gets modified. This is also expected to be more sensitive because it critically depends on the relative detuning of the vibrational modes in both diabatic potentials and . Besides Fig. S9, the spectroscopic data for low field was also used in Fig. 4 B where we discuss the breackdown of the Born-Oppenheimer approximation around the intersection. Because a numerical treatment of the vibronic coupling at finite magnetic field is numerically too challenging due to the additional Zeeman sublevels appearing in the potential calculations, we reduced the field to G. All theoretical calculations in this work were done at zero field where the number of contributing pair states can be significantly increased.
During the experiment, the atoms were pinned in the lattice at lattice depths of for both lattice directions in the atomic plane, where is the recoil energy. For the lattice perpendicular to the atomic plane, we chose a depth of . If not stated otherwise, the UV light was polarized in the atomic plane. For all measurements in this work, for and both polarizations of the UV light, we used a single-photon Rabi coupling of MHz between the ground state and the Rydberg state. In the case of the ground state discussed in Fig. S4, the laser power was reduced in order to compensate a larger Clebsch-Gordan coefficient. As in our previous works Zeiher et al. (2016), was calibrated by Ramsey spectroscopy in the ground state manifold. The detuning was always measured relative to the center of the two Zeeman sublevels.
III.1 Spectroscopic data
The broad spectrum detuned from the Rydberg resonance shown in Fig. 2 A contains roughly data points with frequency spacing of kHz. Each datapoint represents the average atom number of around 10 experimental shots, analyzed in a circular region-of-interest of 80 sites around the center of the cloud. In order to capture all narrow-linewidth macrodimer resonances, the frequency was swept over a range of kHz during the illumination time of ms. The two isolated resonances at the red-detuned side of the spectrum are technical artefacts due to the relative detuning of MHz of the two beams creating our in-plane optical lattice.
As a result, the Zeeman-splitted transitions from to and reappear on both sides of the Rydberg resonance. While the sidebands at the red-detuned side of the resonance are recognized easily, they are overlapped with the macrodimer resonances and on the blue-detuned side. The overall decrease of the coupling strength for even vibrational quantum numbers observed in Fig. 2 A and Fig. 2 B is smaller compared to the actual value due to the strong saturation of the lower macrodimer lines. For the high-resolution spectroscopy of the lowest vibrational level shown in Fig. 2 C, we illuminate the cloud only for ms in order to avoid artificial broadening. Additionally, we did not sweep the UV-frequency because the width of the resonance is larger than the separation of the data points. We also repeated the spectroscopy of the lowest line for lower powers and verified that there is no power broadening. This is not surprising because is smaller than the decay rate which is expected to be at least twice as large as the decay rate of an isolated Rydberg atom in . In Fig. 2 C, one can also recognize a region where the Lorentzian fit deviates slightly from the data. This is reproducible and most likely due to a weakly coupled state originated from the intersecting state , which overlaps with the first strong macrodimer line. For the additional spectroscopic data shown in Fig. 4, Fig. S9 and the left panels of Fig. S7, different data points are separated by kHz and the illumination time was ms. Here, we swept the frequency from kHz around the central frequency in order to avoid missing narrow lines between the data points.
III.2 Correlation measurements
For the correlation measurements, we started with a -filled Mott insulator and illuminated the cloud with UV light resonant with a macrodimer resonance until the filling decreased to . We took about 200 images under the same conditions. Details on the measurements as well as the measured correlation values are shown in Table S2. We verified that the recapture probability of our excited molecules is negligible by measuring only atoms in after preparing a cloud in and shining UV light resonant with the lowest macrodimer line for various times. Then, we perform a push-out beam resonant with the transition from the manifold to the D2 line. We find that the negligible amount of atoms does not increase over time, while the total atom number decreases due to macrodimer excitation. Because Rydberg atoms can decay to both hyperfine ground states, we conclude that all excited macrodimers leave the system as pairs. We also checked that the measured correlations along the strongly coupled diagonal orientation are consistent with a reduction of the filling from the initial value to , which is close to the observed final fillings and indicates that losses are dominated by correlated atom loss along the strongly coupled lattice diagonal. Finally, we have checked that the initial pair loss dynamics from the correlation measurements is consistent with the calculated Rabi coupling kHz to the lowest macrodimer state shown in Fig. S5 via a numerical simulation of the optical Bloch equations.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jones et al. (2006) K. M. Jones, E. Tiesinga, P. D. Lett, and P. S. Julienne, Rev. Mod. Phys. 78 , 483 (2006) . · doi ↗
- 2Regal et al. (2003) C. A. Regal, C. Ticknor, J. L. Bohn, and D. S. Jin, Nature 424 , 47 (2003) . · doi ↗
- 3Moses et al. (2015) S. A. Moses, J. P. Covey, M. T. Miecnikowski, B. Yan, B. Gadway, J. Ye, and D. S. Jin, Science 350 , 659 (2015) . · doi ↗
- 4Liu et al. (2018) L. R. Liu, J. D. Hood, Y. Yu, J. T. Zhang, N. R. Hutzler, T. Rosenband, and K.-K. Ni, Science 360 , 900 (2018) . · doi ↗
- 5Gallagher (1994) T. F. Gallagher, Rydberg Atoms , Cambridge Monographs on Atomic, Molecular and Chemical Physics (Cambridge University Press, 1994). · doi ↗
- 6Bendkowsky et al. (2009) V. Bendkowsky, B. Butscher, J. Nipper, J. P. Shaffer, R. Löw, and T. Pfau, Nature 458 , 1005 (2009) . · doi ↗
- 7Shaffer et al. (2018) J. P. Shaffer, S. T. Rittenhouse, and H. R. Sadeghpour, Nature Communications 9 , 1965 (2018) . · doi ↗
- 8Miller et al. (1993) J. D. Miller, R. A. Cline, and D. J. Heinzen, Phys. Rev. Lett. 71 , 2204 (1993) . · doi ↗