Development of a New Parameter Optimization Scheme for a Reactive Force Field (ReaxFF) Based on a Machine Learning Approach
Hiroya Nakata, Shandan Bai

TL;DR
This paper introduces a machine learning-based method to optimize ReaxFF parameters for reactive molecular dynamics, enabling accurate high-temperature simulations and insights into chemical vapor deposition processes.
Contribution
A novel parameter optimization scheme combining k-nearest neighbors and random forest algorithms for ReaxFF, improving efficiency and accuracy in high-temperature reactive MD simulations.
Findings
Optimized ReaxFF parameters accurately predict high-temperature properties.
The method successfully simulated crystal growth in CVD of α-Al₂O₃.
Surface growth behaviors were elucidated using the new parameters.
Abstract
Reactive molecular dynamics (MD) simulation is performed using a reactive force field (ReaxFF). To this end, we developed a new method to optimize the ReaxFF parameters based on a machine learning approach. This approach combines the -nearest neighbor and random forest regressor algorithm to efficiently locate several possible ReaxFF parameter sets, thereby the optimized ReaxFF parameter can predict physical properties even in a high-temperature condition within a small effort of parameter refinement. As a pilot test of the developed approach, the optimized ReaxFF parameter set was applied to perform chemical vapor deposition (CVD) of an -AlO crystal. The crystal structure of -AlO was reasonably reproduced even at a relatively high temperature (2000 K). The reactive MD simulation suggests that the (110) surface grows faster than the…
Click any figure to enlarge with its caption.
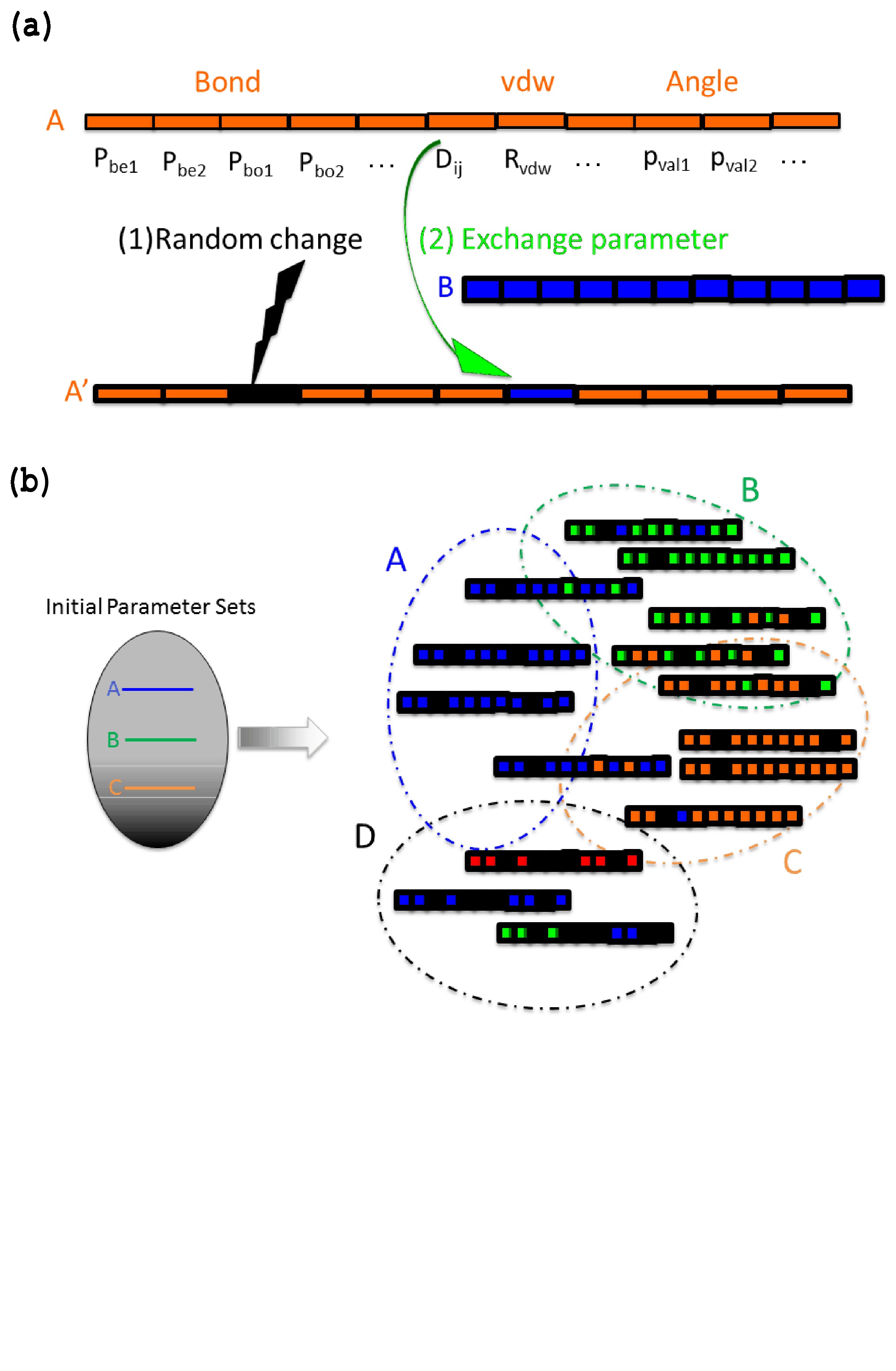 Figure 1
Figure 1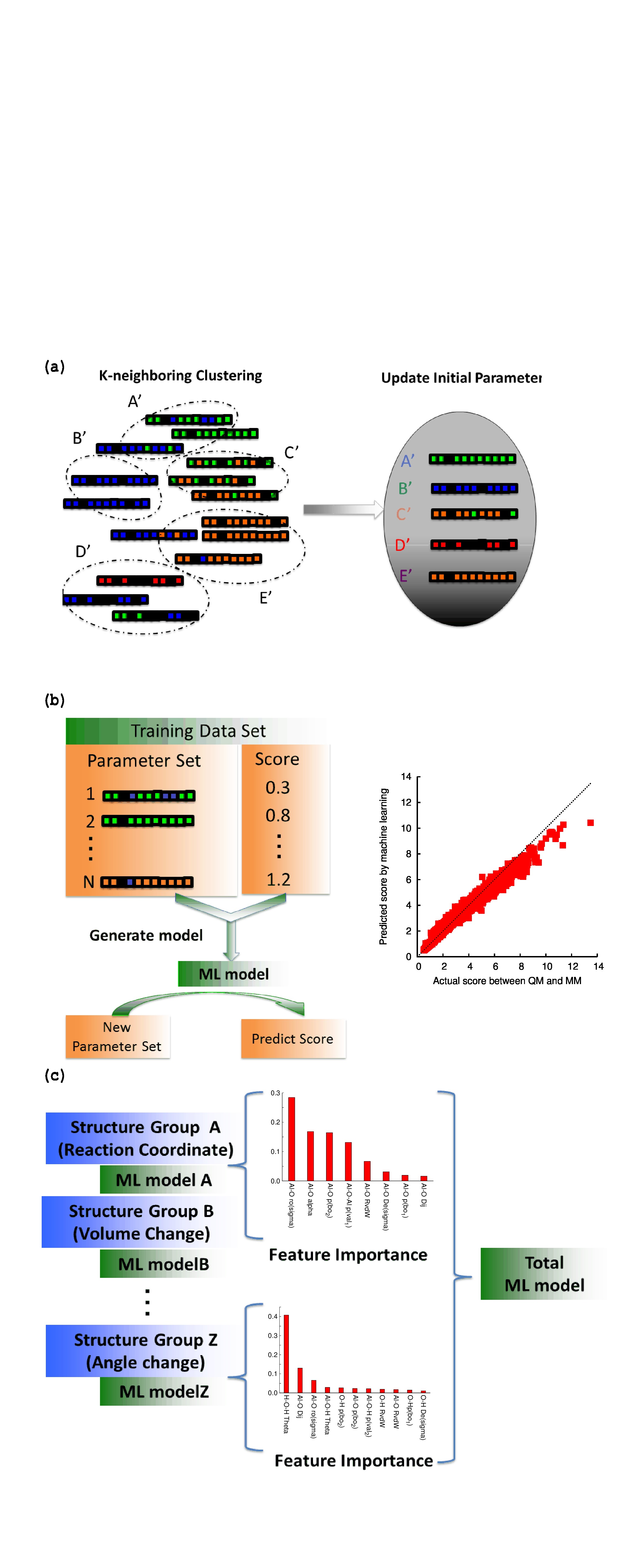 Figure 2
Figure 2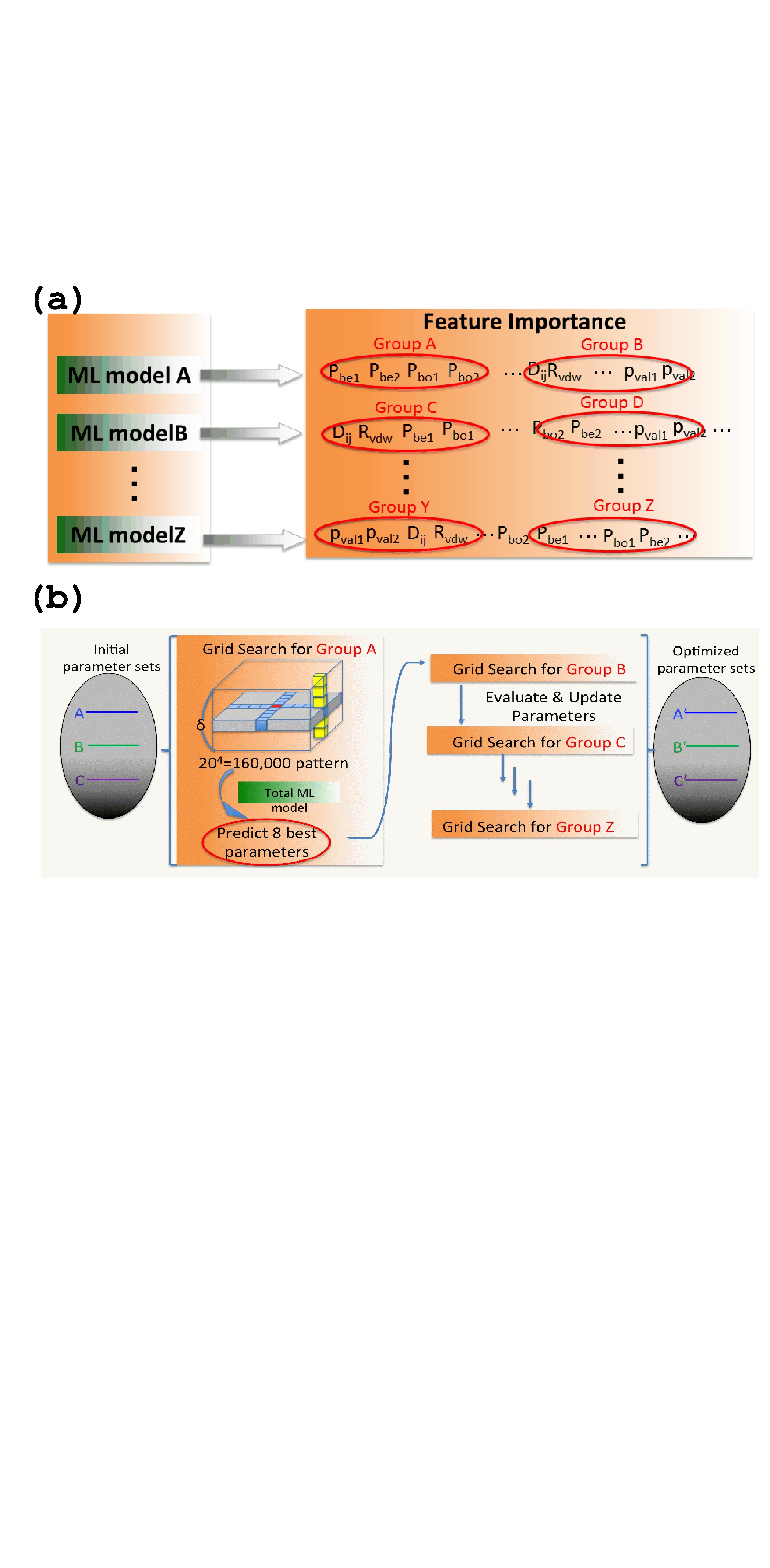 Figure 3
Figure 3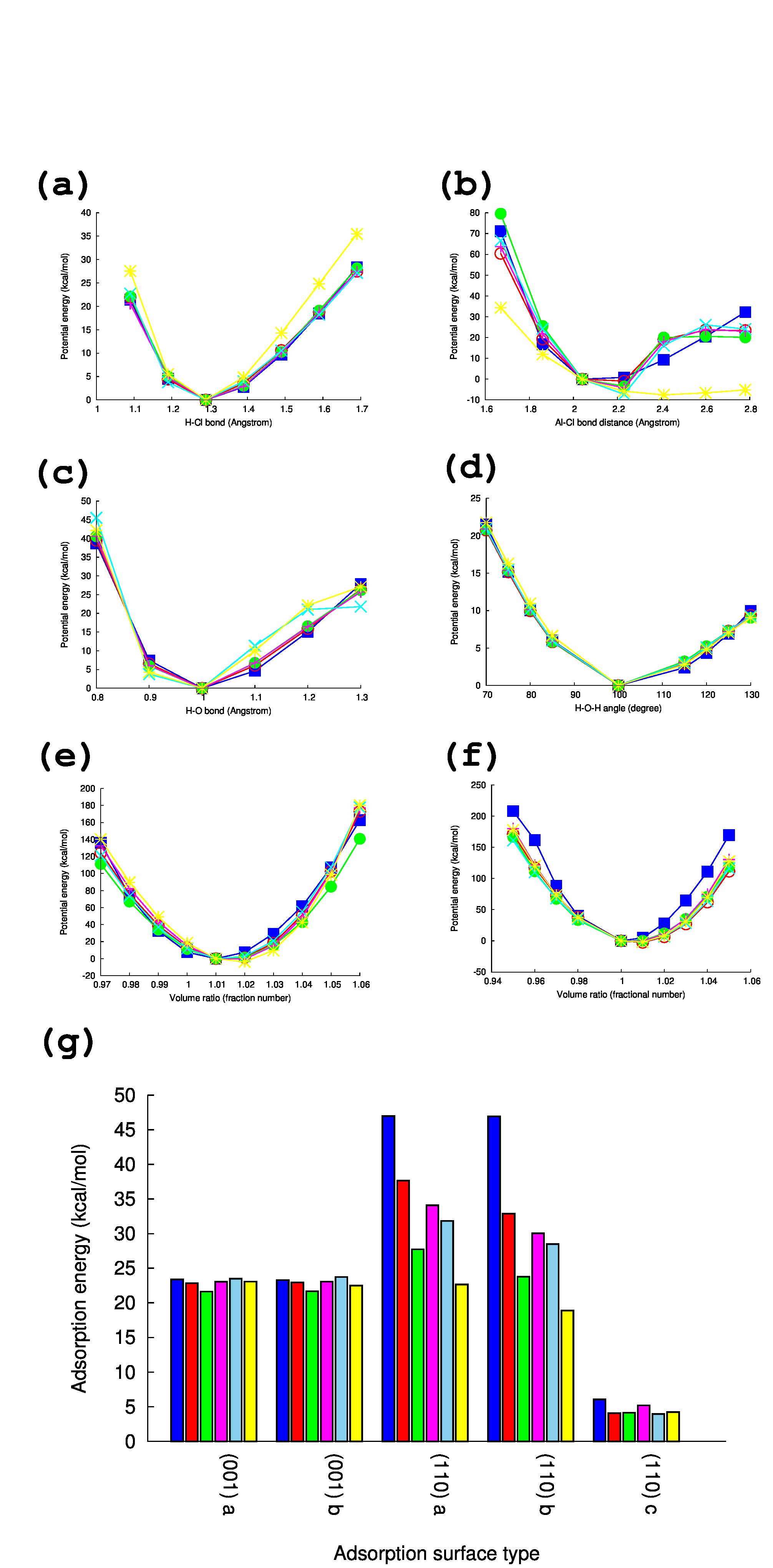 Figure 4
Figure 4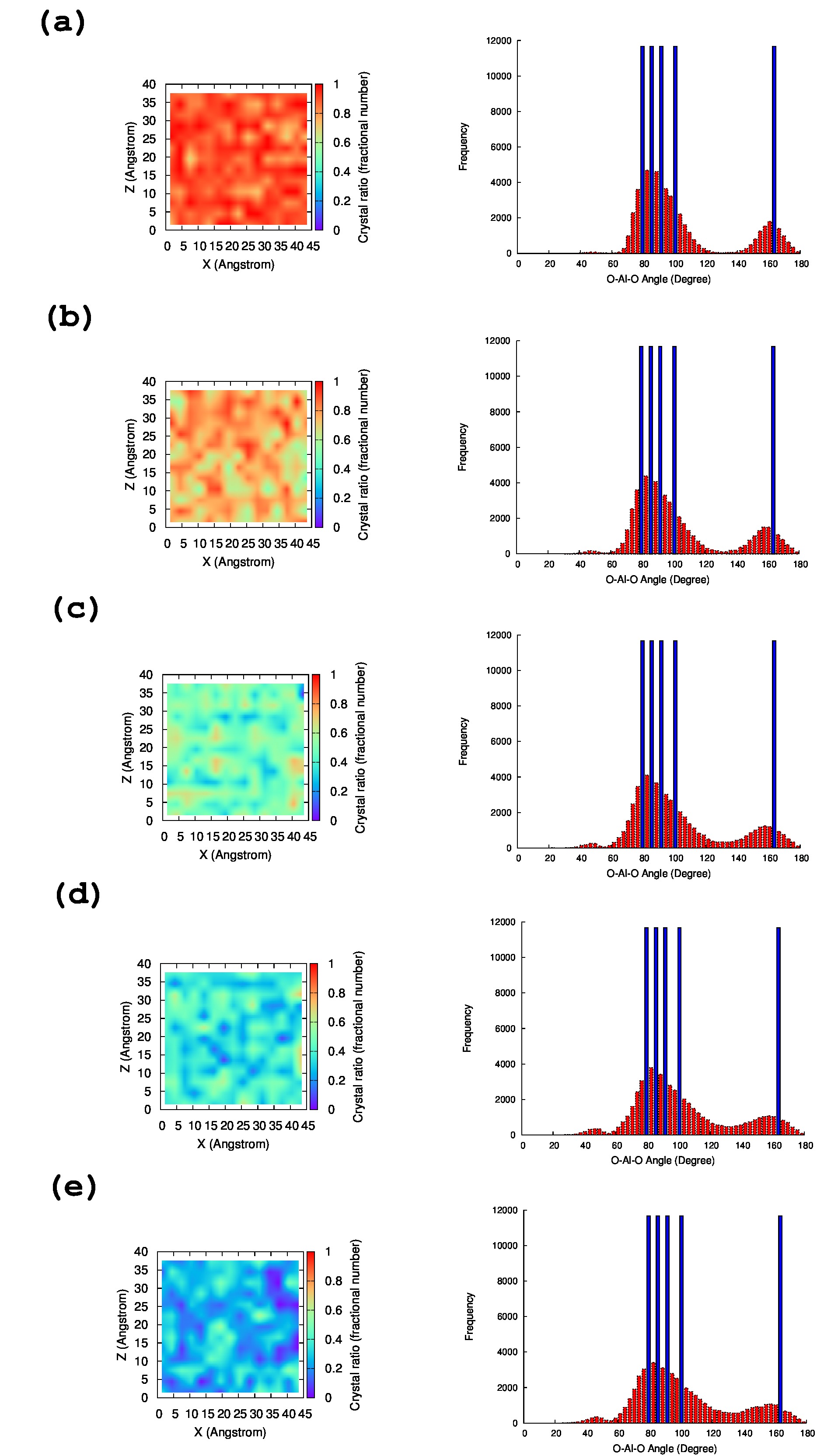 Figure 5
Figure 5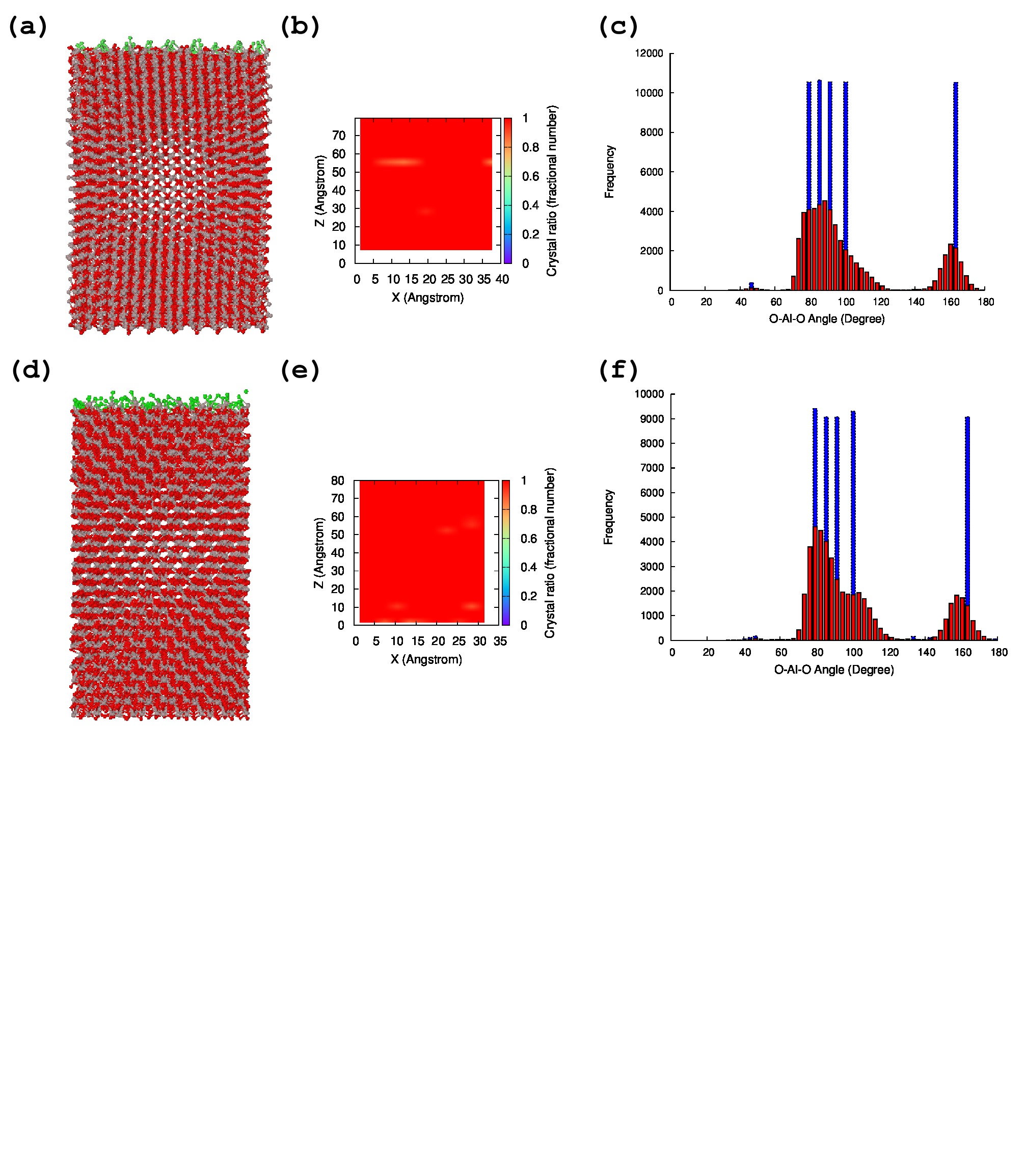 Figure 6
Figure 6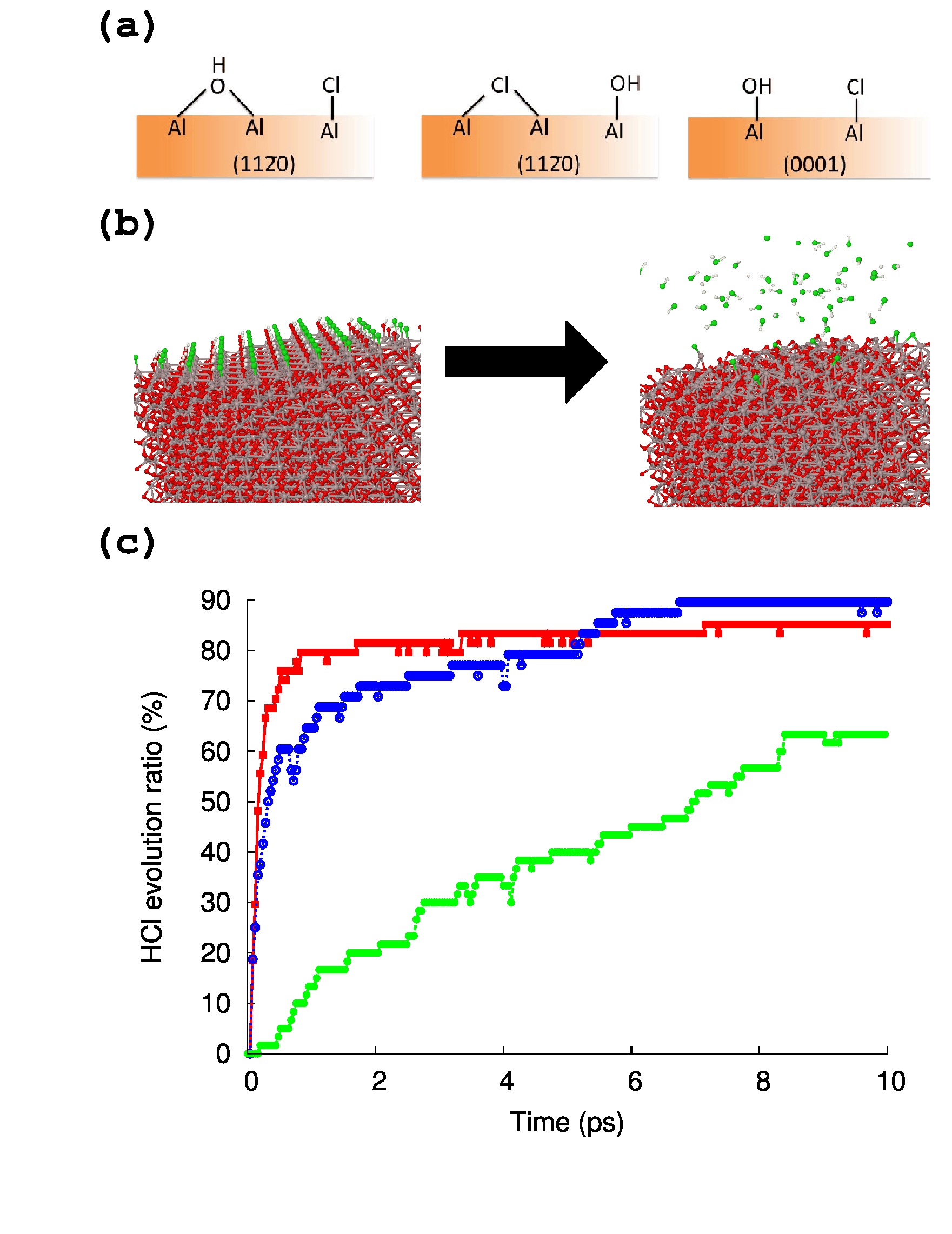 Figure 7
Figure 7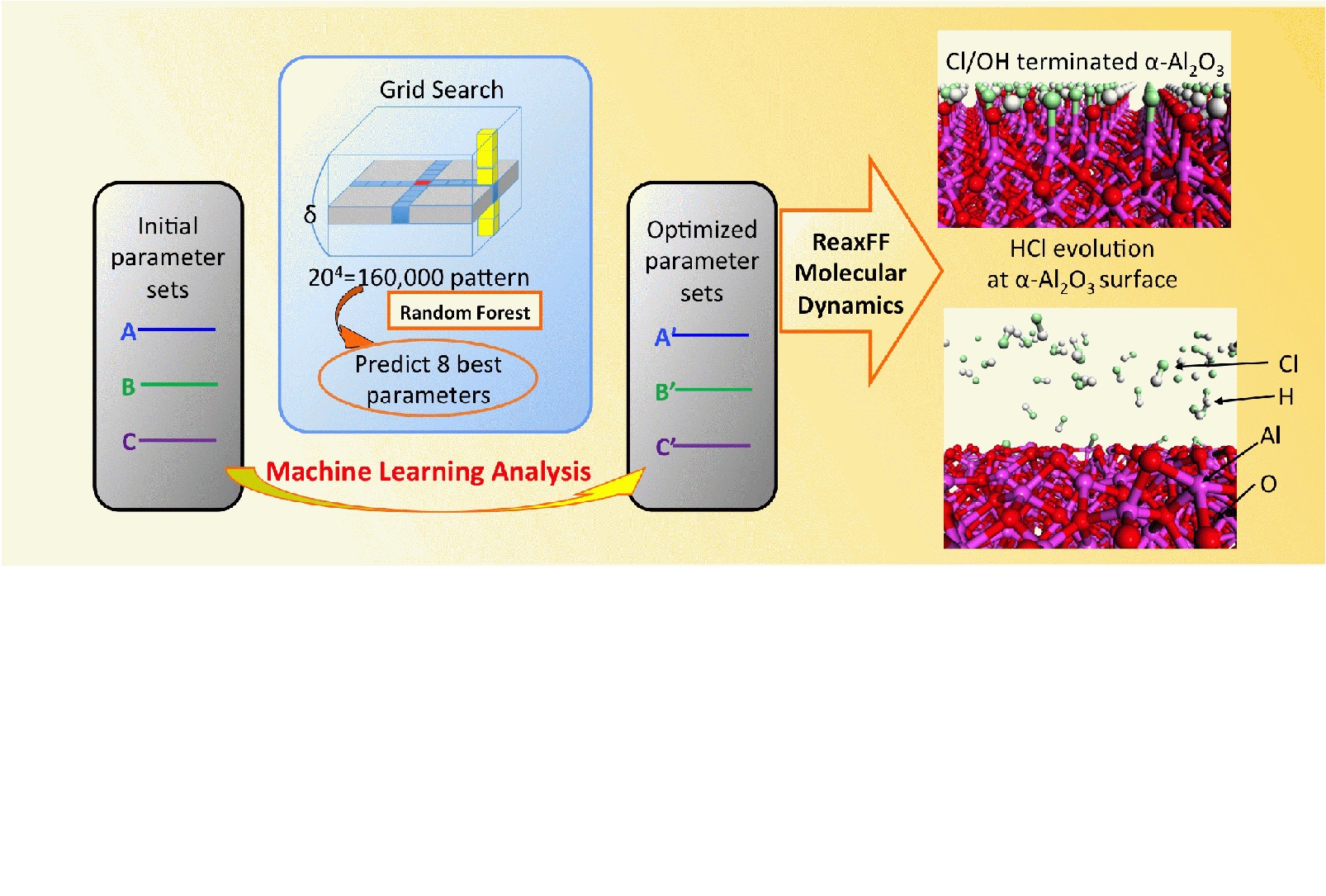 Figure 8
Figure 8| Label | rmse | MaxErr |
|---|---|---|
| A | 0.100 | 0.577 |
| B | 0.107 | 0.840 |
| C | 0.114 | 0.495 |
| D | 0.123 | 0.586 |
| E | 0.173 | 0.583 |
| F | 0.091 | 0.677 |
| G | 0.138 | 0.579 |
| H | 0.104 | 0.536 |
| Label | ratio of six coordination |
|---|---|
| A | 89.69 |
| B | 58.92 |
| C | 49.33 |
| D | 37.47 |
| E | 25.87 |
| F | 48.51 |
| G | 62.91 |
| H | 59.57 |
| parameter type | A | B | C | D | E | F | G | H | std/avg |
|---|---|---|---|---|---|---|---|---|---|
| De | 121.3105 | 102.9408 | 100.976 | 91.03454 | 94.02349 | 95.02255 | 99.82974 | 100.2807 | 8.610 |
| p(bo1) | -0.0581 | -0.0692 | -0.0578 | -0.0661 | -0.0705 | -0.0605 | -0.0598 | -0.0708 | -8.237 |
| p(bo2) | 7.9659 | 7.6978 | 8.9451 | 8.2364 | 9.9658 | 8.3418 | 8.6086 | 8.0779 | 7.863 |
| Dij | 0.0643 | 0.0868 | 0.0678 | 0.0888 | 0.0667 | 0.0937 | 0.0719 | 0.0640 | 15.125 |
| RvdW | 1.7782 | 1.7295 | 1.7959 | 1.8925 | 1.8529 | 1.7075 | 1.7065 | 1.7036 | 3.820 |
| alfa | 11.1010 | 11.1040 | 11.161 | 10.159 | 10.881 | 11.116 | 11.181 | 11.683 | 3.596 |
| ro(sigma) | 1.5421 | 1.5707 | 1.5915 | 1.572 | 1.6638 | 1.5666 | 1.5846 | 1.5703 | 2.117 |
| p(val1) | 28.7692 | 33.6726 | 11.134 | 31.3518 | 32.8224 | 34.7920 | 30.8857 | 33.9987 | 24.403 |
| p(val2) | 1.4601 | 1.1855 | 1.6583 | 0.8882 | 1.1641 | 1.1993 | 1.4369 | 1.2613 | 17.068 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMachine Learning in Materials Science · Semiconductor materials and devices · Thermal properties of materials
Development of a New Parameter Optimization Scheme for a Reactive Force Field (ReaxFF) Based on a Machine Learning Approach
Hiroya Nakata
R and D Center Kagoshima, Kyocera corporation, 1-4 Kokubu Yamashita-cho, Kirishima-shi, Kagoshima, 899-4312, Japan
the authors equally contribute in this study
Shandan Bai
R and D Center Kagoshima, Kyocera corporation, 1-4 Kokubu Yamashita-cho, Kirishima-shi, Kagoshima, 899-4312, Japan
the authors equally contribute in this study
Abstract
Reactive molecular dynamics (MD) simulation is performed using a reactive force field (ReaxFF). To this end, we developed a new method to optimize the ReaxFF parameters based on a machine learning approach. This approach combines the -nearest neighbor and random forest regressor algorithm to efficiently locate several possible ReaxFF parameter sets, thereby the optimized ReaxFF parameter can predict physical properties even in a high-temperature condition within a small effort of parameter refinement. As a pilot test of the developed approach, the optimized ReaxFF parameter set was applied to perform chemical vapor deposition (CVD) of an -Al2O3 crystal. The crystal structure of -Al2O3 was reasonably reproduced even at a relatively high temperature (2000 K). The reactive MD simulation suggests that the (110) surface grows faster than the (0001) surface, indicating that the developed parameter optimization technique could be used for understanding the chemical reaction in the CVD process.
I Introduction
A chemical reaction that typically involves bond cleavage and formation plays an important role in the field of material science. In principle, it is possible to handle the chemical reaction by a quantum mechanical (QM) approach, but the simulation of the chemical reaction is still computationally challenging. Because expensive Hessian calculationshess1 ; hesspar or molecular dynamics (MD) simulations are needed for simulation of the chemical reaction, the QM applications are limited to within only 100 or 200 atoms in the system, and a large-scale (more than 10,000 atoms) reaction analysis is virtually impossible within the framework of the standard QM approach.
Two approaches are currently possible for a large-scale reaction analysis based on the QM method: a hybrid quantum mechanical/molecular mechanical (QM/MM) approachQMMM1 ; QMMM2 ; ONIOM and a fragment-based approach.frgrev ; MCF ; DC0 ; XPol ; DC2 ; DC3 ; eemb2 ; MFCC3 ; PMISP ; ADMA ; EFP2013 ; pdf1 Although the majority of MD studies of large-scale reaction analysis employ the QM/MM approach, the boundary conditions between QM and MM are sometimes a problem. Thus, MD based on QM/MM is not straightforward, and a challenging study of adaptive QM/MM molecular dynamics was recently reported.QMMMadaptive ; watanabe2016adaptive In contrast, in the fragment-based approach, vibration analysis has been developedIMCMO ; GEBFHESS ; SMFHESS ; MTAhess ; QMQM ; FMO_HESS and a 10,000-atom enzyme reaction has become possible.FMOFD2
However, despite the above success of the QM-based approach, MD simulations with QM are limited to within only 1100 ps.FMOMD Thus, it is not yet practical to apply QM-based MD to a chemical reaction in a material in which the system size is typically of nanometer scale and nanosecond simulation time.
One possible way to investigate the chemical reaction in nanoscale systems is currently the classical force field approach.van2001reaxff ; EVBforce Because the classical force field approach has difficulty in handling the chemical reaction, the reactive force field (ReaxFF) approach was developed by van Duin et al.van2001reaxff An additional advantage of ReaxFF is the ability to study a wide range of material simulations, including solid-state crystal, molecular crystal, and gas molecules. Thus, ReaxFF has been applied to many chemical reactions in materials.reaxff01 ; reaxff02 ; reaxff03 ; reaxff04 ; reaxff05
To apply the ReaxFF MD simulation of a chemical reaction, force field parameter refinement is sometimes necessary. The force field parameter fitting is usually performed to reproduce the static properties such as energy, force, and charge by using a particular QM training data set. To fit the ReaxFF parameters to the QM training data set, a genetic lgorithm,reaxFFGA01 ; reaxFFGA02 ; reaxFFGA03 ; reaxFFGA04 ; reaxFFGA05 and a multiobjective genetic algorithmGARFF ; ReaxFFGAM have been intensively studied; thus, force field parameter optimization based on the QM method has become relatively easy.
The success of the ReaxFF simulation and force field fitting has opened up new possibilities for application in computational material science. However, it is not yet straightforward to apply ReaxFF to a new chemical reaction, in particular in a nonequilibrium process such as chemical vapor deposition (CVD). This is because, in a nonequilibrium process, there is uncertainty in the quantification of simulated quantities of interest, and the preparation of a QM training data set is sometimes impossible for our quantities of interest. Mishra et al. improved the uncertainty quantification by including the dynamic approach to simulating reactive MD.mishra2018multiobjective However, even with the above success of multiobjective parameter refinement including the dynamic properties, parameter fitting takes a considerable time depending on the properties in which we are interested. For example, if during a particular reaction a free energy barrier or infrared (IR) spectroscopy is required, parameter refinement is necessary for these physical properties. However, this involves a considerable computational cost, and it is sometimes impossible within our available computer resources; thus, parameter optimization for a nonequilibrium process is still a challenging issue.
Thus, force field parameter refinement in ReaxFF warrants further investigation. This study focuses on how to efficiently obtain an appropriate parameter set for the physical properties of interest. For this purpose, a new multiobjective parameter fitting process was developed. The distinct features in the proposed method are as follows: (i) efficiently obtaining several local minima during the parameter optimization step, (ii) an efficient optimization process based on an ML approach, and (iii) transferability to other parameter optimization processes given the simple structure and easy implementation.
In this study, the proposed ReaxFF parameter optimization method is first described in detail. Then, the parameter optimization results are briefly described. As a pilot test, a ReaxFF parameter force field for Al2O3 is generated, and the performance of the reactive MD simulation is evaluated. Al2O3 is used for many important industry materials such as passivation films for solar cells and polishing materials for cutter grinders. The Al2O3 crystal is usually applied as a coating in a nonequilibrium process such as CVD or atomic layer deposition (ALD). Because it is necessary to perform the simulation at a high temperature, reproduction of the crystal structure even at a high temperature is important for a meaningful simulation. As a pilot test of parameter optimization, the MD simulation was performed for the bulk and surface crystal structure to demonstrate the effectiveness of the proposed parameter-fitting approach. Finally, the reactive MD simulation was performed for the pilot test.
II ReaxFF Parameter Optimization Using ML
The proposed parameter optimization scheme comprises three important steps. (i) The predefined initial parameters are randomly modified to generate the training data set for ML (Figure 1). (ii) Data analysis based on ML is performed for this training data set (Figure 2). (iii) A grid search parameter optimization based on the ML model is performed (Figure 3). The respective steps are denoted as (i) random parameter sampling, (ii) data analysis based on ML, and (iii) grid-search parameter optimization. A detailed description of each step follows below.
II.1 Definition of parameter sets and random parameter sampling
The potential energy in ReaxFF is
[TABLE]
where and are the potential energies for bond and lone pairs, respectively; and are the energy penalties for over- and undercoordination; is the valence angle term; is the penalty energy term related to the double bond; is the energy of the three-body conjugation term; is the energy of the strong triple bond; is the potential energy for torsion; is the energy of hydrogen bonds; and and are the van der Waals and Coulomb interaction energies, respectively (for detail see van2001reaxff ).
Thus, the ReaxFF parameter set contains the information of bonds (, , , , etc.), van der Waals force ( and , etc.), and angle related to three-body parameters (, ). The parameters (, , , , ) are defined as a set of parameters for MD simulation, and these ReaxFF parameters are denoted as the parameter set.
Two different types of random change are introduced to generate a variety of types of parameter sets, as follows (Figure 1-(a)). One is a random change in the parameter set (Figure 1-(a)), and several parameters are selected () randomly with a predefined probability =0.5, where is the initial parameter value, such as or . The new randomly changed parameter is
[TABLE]
where is
[TABLE]
where and are scale factor and random number, respectively. In this study, and are used; therefore, the original parameter (ptype) can be changed by 10%. Another type of parameter modification involved exchanging the parameter value for a value in another initial parameter set with a probability , and 0.2 is used for the probability of exchange parameter sets in this study.
This modification in the parameter set is inspired by the genetic algorithmreaxFFGA01 and efficient sampling of various kinds of parameter sets is possible. As an example, a schematic illustration of parameter sampling is shown in Figure 1-(b). In Figure 1-(b), the different colors denote the different kinds of initial parameter sets, and three initial parameter sets are shown in Figure 1-(b). By performing the random sampling according to Figure 1-(a), the parameter set spans over the space in the initial parameter sets of A, B, and C, as shown on the left-hand side of Figure 1-(b). In addition, a new sampling space is sometimes found by the random change of the initial parameter sets of D, as shown on the right-hand side of Figure 1-(b); thereby, efficient parameter sampling becomes possible.
II.2 Evaluation of the ReaxFF parameter sets
The previous section introduced the random parameter sampling to obtain the training data set (ReaxFF parameter sets). For the data analysis based on ML, each training data set (ReaxFF parameter set) should have a score that represents the validity of the data set.
The score for each training ReaxFF parameter set can be evaluated as follows:
[TABLE]
where is the number of geometry sets, and each geometry set contains different structures that represent a particular potential energy curve or physical properties such as the potential energy along the volume change in the -Al2O3 crystal or the bond cleavage of H2O. and are the score and weight for each geometry set , and we used the root mean square error (rmse) to evaluate . and are quantities evaluated with ReaxFF and QM for each th structure in the geometrical set . It is possible to use any kind of physical properties for the quantities and , and we used the potential energy in this study.
Because the absolute value of is significantly different between the respective geometry sets , and were standardized:
[TABLE]
where and are the average and standard deviation (SD) for the geometry set of . The standardized quantities are used in eq 5 instead of the original values .
II.3 Data analysis based on ML
Now the training data set contains the parameter set , the total score , and the scores for each geometry set (for each different kind of property). By using this training data set, three different kinds of data analysis were performed based on the ML approach: (a) update the initial parameter set by the -nearest neighbor algorithm (Figure 2-(a)), (b) generate the ML model (Figure 2-(b)), and (c) extract the feature importance (Figure 2-(c)).
In step (a), the initial parameter set is updated by the -nearest neighbor algorithm (Figure 2-(a)). As shown in Figure 2-(a), the -nearest neighbor algorithm separates the training data sets into groups, and the -nearest neighbor algorithm makes the distance between the groups as large as possible. In this study, eight was used for , and increasing improves the convergence of parameter optimization, but the computational time increases because of the following grid search optimization (see next section). Thus, the number of between six and eight seems an appropriate choice for parameter . From each group classified by the -nearest neighbor algorithm, the parameter set that has the lowest total score is selected, and the initial parameter sets are updated as shown in Figure 2-(a).
Figure 2-(b) is a schematic illustration of the ML approach. The ML model was constructed so as to reproduce the score ( or ) from the parameter sets () (see the right-hand side of Figure 2-(b)). In this study, the training data set contained the total score , and for each geometry set ; therefore, for + 1, an independent ML model can be constructed. To make ML models, a random forest regressor model was used in this study. One of the merits of using the random forest regressor is the feature importance of the by-product, which describes the sensitivity of respective elements in the parameter set.
The entire structure of the ML model is depicted in Figure 2-(c). The training data set contains a score ( ) for each geometry set ( different physical properties), and each geometry set has an ML model and feature importance. It is also possible to construct an ML model for the total score (). The grid search parameter optimization was performed using the ML model for the total score and each geometry set, as described below.
II.4 Grid search parameter optimization
The reaming process for ReaxFF parameter optimization was the grid search parameter optimization based on the ML model generated as described in the previous section. The schematic illustration for grid search parameter optimization is shown in Figure 3.
Each geometry set ( different physical properties) contains the independent ML model, and distinct types of feature importance. According to the feature importance, the parameter set is split into several groups, and each group contains four parameters. For example in Figure 3-(a), ML model A contains the parameters (, , , , , , , , , ). The four parameters (, , , ) are the most important parameters for geometry set A, and this four-parameter set was denoted as Group A. Likewise, the four less important parameters such as (, , , ) were denoted as group B. Because each ML model contains distinct feature importance levels, the respective groups (A, B, , Z) contain different combinations of parameters.
Then the grid search parameter optimization is performed for each group (A, B, , Z) independently, and the summary of the grid search parameter optimization is shown in Figure 3-(b). First, the initial preliminary parameter sets were prepared according to Figure 2-(a). To perform grid search parameter optimization, the initial parameters span around their initial values, and the range is split into 20 grid points. As each group contains four parameters, the total number of grid points is , and the direct evaluation of the total score in eq 4 is time-consuming. The ML model can predict the total score within negligible computational time; therefore, the computational time can be significantly reduced.
Using the ML model, eight best parameter sets were predicted from the 160,000 different parameter sets, and the total scores were evaluated only for these eight parameter sets using eq 4. If the evaluated total score is less than the score in the initial parameter set, the initial parameter set is updated. The grid search parameter optimization procedure is iteratively performed for each group A, B, , Z (Figure 3).
II.5 Summary of parameter optimization based on the ML approach
The previous sections described the respective parts of parameter optimization; the purpose of this section is to connect the respective steps to a cycle of parameter optimization.
The random parameter sampling illustrated in Figure 1 plays two important roles. The random sampling can escape trapping a local minimum during parameter optimization, and could efficiently sample the training data set for the ML model.
The ML model was constructed based on the training data set, and involves three important roles. First, the -nearest neighbor algorithm selects each initial parameter set as far as possible, providing efficient finding of possible local minima. Second, the random forest regressor model can efficiently predict the parameter set containing the lowest total score, and significantly reduce the computational cost by avoiding the evaluation of many redundant parameter sets. Third, the feature importance estimated by the random forest regressor provides a guide for the optimization of the respective parameters, and the groupwise grid search can be effective.
By iteratively performing random structure sampling, ML analysis, and grid search parameter optimization, we can obtain possible local minima, which contain different uncertainties for the other physical properties, such as crystal structure at high temperature. In the following discussion, the effectiveness of this approach for performing an MD simulation is demonstrated.
III Computational Detail
The computational models used for potential energy evaluated by QM were based on the crystal structure of Al2O3. The geometry sets for the quantity evaluation in eq 5 were the potential energy curve along the volume change in -Al2O3 crystal structure, the volume change in -Al2O3 crystal structure, the bond cleavage in H2O, HCl, and AlCl3, the angle potential energy curve for H2O, and the absorption energy for H2O on the (0001) and (110) surface of -Al2O3.
A plane wave-based density functional theory (DFT) program Castep was usedCASTEP1 ; CASTEP2 with the Perdew, Burke and Ernzerhof (PBE) functional.PBE1 ; PBE2 All the calculations were performed with ultrasoft core potentialsultrasoft generated on the fly, and the cutoff energy was set to 571.4 eV. The electronic configurations of the atoms were Al:3s2 3p1, O:2s2 2p4, H:1s1, and Cl:3s2 3p5. We adopted the convergence criterion of 0.03 eV/A for geometry optimizations, and 0.0001 eV for the self-consistent field calculations of electronic states. A 2 2 2 uniform mesh for k-space integrations was used for all the bulk simulations, and a 2 2 1 mesh was used for the surface models.
To evaluate the validity of the obtained ReaxFF parameter set, MD simulations were performed for the crystal structure of -Al2O3. The system size was 42.8 49.45 38.973 and the total number of atoms was 9720. The coordination number (CN) of Al atoms and the angle between O–Al–O were evaluated regardless of whether or not the crystal structure was maintained during the MD simulation even in the relatively high-temperature region (2000 K).
To show the feasibility of the optimized ReaxFF parameter set, the MD simulations were also performed with surface structures. Two kinds of surface structure, (0001) and (110), were evaluated. A previous study of thermodynamic simulationsAl2O3Firstprinciple ; CVDAl suggests that the -Al2O3 surface is terminated by Cl; therefore, the simulation models were also terminated by Cl, and the crystal structure was investigated. The system size for the surface model was 32.97 33.313 60.625, and the total number of atoms was 7952. For the pilot test of the ReaxFF parameter, a reactive MD simulation was performed using the surface model of -Al2O3, and the HCl evolution reaction was evaluated.
All the MD simulations in bulk structures were performed at 2000 K, which nearly corresponds to the melting point of the -Al2O3 crystal. The MD simulations on the surface model were performed at 1223 K, because the CVD experiments on -Al2O3 are typically conducted at 1223 K. For the MD simulation of bulk and surface structures, NVT MD simulation was performed with a Nose–Hoover thermostat, and the velocity Verlet integrator was used for time integration, and the time step was 0.1 fs. The number of time steps was 100,000, which corresponds to the 10 ps simulation time.
IV Results and Discussion
IV.1 Parameter fitting
The rmse and maximum errors (MaxErr) between the ReaxFF parameter and the QM potential energy are summarized in Table 1. The rmse and MaxErr were normalized by the SD (see eq 6). As shown in Table 1, all the rmse values estimated by the ReaxFF parameter set are around 0.1, and the parameters could reproduce the QM result within 35% errors. Thus, almost all the optimized parameter sets represent a reasonable agreement with the QM calculation (see below for more detail in the comparison of the potential energy between QM and the ReaxFF parameter).
The point of the proposed approach is that the single ReaxFF parameter optimization task generates possible local minima; therefore, independent ReaxFF parameter sets could be obtained directly. To show the difference between the optimized parameter sets, a summary of the respective parameter sets is shown in Table 3. A significant difference is observed between the parameter sets in terms of -Al2O3 crystal structure; the parameters related to Al and O are shown in Table 3. As shown in Table 3, the differences between the parameters are relatively large, especially the angle-related parameters p(val1) and p(val2), which differ by around 24% (std/avg) and 17% (std/avg), respectively. It is interesting to obtain these different kinds of ReaxFF parameters with the power of the ML technique.
The parameter fitting results (AH in Table 1) in terms of the respective structure data sets are shown in Figure 4 for AE, and the Supporting Information (SI) for FH. (The MD simulation results with parameter sets FH present similar results to AE, and the results are shown in the SI.) The comparison between the QM calculations (blue filled squares) and the ReaxFF parameter results shows good agreement with each other. The extent of errors shown in Figure 4 is often found in other ReaxFF parameter optimizations,ReaxFFQMvsMM01 ; ReaxFFQMvsMM02 ; ReaxFFQMvsMM03 ; ReaxFFQMvsMM04 ; ReaxFFQMvsMM05 ; ReaxFFQMvsMM06 ; ReaxFFQMvsMM07 which suggests that the optimized parameter set was reasonable for reactive MD simulation. The next section presents how the parameter sets AH affect the crystal structures during the MD simulations at high temperature (2000 K).
IV.2 Evaluation of the parameter sets using MD in -Al2O3
To show a specific example for the difference in physical property depending on the parameter sets AH, MD simulations were performed with -Al2O3 for each parameter set, and we investigated whether the crystal structure of -Al2O3 was maintained or not. For this purpose, the CN of Al was calculated. The Al atom possesses six bonds with O, therefore the CN is six. If the crystal structure of -Al2O3 is maintained, all the CNs of Al should be six, so that the ratio of CN six of Al is evaluated to determine the crystalline structure during the MD simulation. For simplicity, we denote the ratio of CN 6 of Al as the crystal structure ratio.
The results of MD simulation using the respective optimized parameter set are summarized in Table 2. As shown in Table 2, a significant deviation of the crystal structure ratio is observed between the parameter sets AH from 25% to 89%. The crystal structure of -Al2O3 is maintained for about 90% in the MD simulation using with parameter set A. In contrast, with parameter set E, most of the Al atoms deviate significantly, and the crystal structure ratio is only 25.87%.
To investigate further the crystal structure ratio at 2000 K, Figure 5 presents a graphic illustration of the crystal structure ratio, and the O–Al–O angle using the parameter sets AE (see the SI for F and G). The results using parameter set A mostly maintain the crystal structure, and the red color in Figure 5 sharply decreases from parameter sets A to E. Instead, the blue color increases systematically from A to E, which denotes that the geometry obtained by parameter set E becomes amorphous. Likewise, the angle between O–Al–O broadens toward parameter sets A to E. For parameter set A, the angle is mostly around the crystal structure at 0 K, but the distribution of the angle deviates significantly in the MD simulation using parameter set E. With the analysis of CN and the distribution of the O–Al–O angle, we can conclude that parameter set A is the best choice for performing the reactive MD simulation.
As shown in the above example of parameter sets, the well-fitting parameters (e.g., parameter set F) are sometimes not an appropriate choice, because the training structure data set always contains an uncertainty for a particular physical property (in this case, the crystal ratio at 2000 K). One of the possible solutions is increasing the number of training structure data sets; then, the global minimum should be the reasonable parameter set. However, preparing an adequate training structure data set requires expert experience, and it is usually difficult to know how many training structure data sets are actually necessary. Therefore, this approach to find only one global minimum of a ReaxFF parameter requires iterative trial and error to perform a reactive MD simulation.
The approach in this study can reduce the complex procedure for parameter optimization by trying to find different local minima. The -nearest neighbor algorithm separates the data set as far as possible. Therefore, the local minima differ from each other. Consequently, the parameter sets likely contain different kinds of uncertainty for physical properties (Figure 5). In this case, parameter set A provides excellent performance for reproducing the crystal structure at 2000 K.
IV.3 Reactive MD simulation on the -Al2O3 surface
The reactive MD simulation was performed using the optimized parameter set A. The CVD experiment and theoretical thermodynamic study reported that the surface structure of -Al2O3 is terminated by ClAl2O3Firstprinciple ; CVDAl and the HCl evolution step is an important process in the CVD experiment. Therefore, the HCl evolution reaction was investigated in this study.
For this purpose, the surface structures of -Al2O3 (0001) and (110) were investigated using the optimized parameter set A. The result of the MD simulation of the surface structure is shown in Figure 6. In Figure 6, two different kinds of surface structure are shown. The surface structures of -Al2O3 (0001) are shown in Figure 6-(a)–(c), and the surface structures of -Al2O3 (110) are shown in Figure 6-(d)–(f). As shown in Figure 6, the crystal structure is reasonably maintained during the 10 ps MD simulation, and most of the Al atoms have six coordination between O atoms. In addition, the angle O–Al–O shows reasonable agreement with the experimental crystal structure. Therefore, MD simulation on the surface structure is possible.
As the MD simulation on the surface is possible using parameter set A, the HCl evolution reaction was investigated by reactive MD simulation. For this purpose, surface models for both (0001) and (110) were prepared as shown in Figure 7-(a). On the (0001) surface, there is only one type of site, and each site is occupied by Cl or OH (this model is denoted as (0001)). This model contains excessive OH compared with the experiment to accelerate the HCl evolution reaction. Likewise, the (110) surface model is shown in Figure 7-(a), and two different site types are observed on the (110) surface. Thus, two different types of surface structures were evaluated. One is the model that contains OH bridged between two Al atoms, and Cl attached on one Al atom (Figure 7-(a), this model is denoted as (110)-(a)). The other is a model that contains Cl bridged between two Al atoms, and OH attached on one Al atom (this model is denoted as (110)-(b)). In total, three independent reactive MD simulations ((001), (110)-(a), and (110)-(b)) were performed.
The initial and final structures after the 10 ps simulation are shown in Figure 7-(b), and the HCl evolution reactions during the MD simulations are depicted in Figure 7-(c). Significant differences are observed between the three surface models. Most of the HCl molecules immediately evolve at 1223 K in the simulation model (0001) and (110)-(a), whereas the evolution rate of HCl in (110)-(b) is significantly slower than the others. This slow HCl reaction rate in (110)-(b) is caused by the tightly binding Cl atoms on the two Al atoms; the final HCl evolution reaction ratio on (110)-(b) is only 63.33%. In contrast, most of the HCl molecules evolve in the (0001) model in the initial 3 ps, but the HCl evolution reaction on the (0001) surface stops at 85.19% (HCl:ClAl = 8:2), whereas the HCl evolution on the (110)-(a) surface continues for 10 ps, and the final HCl evolution ratio is 89.58%.
The rate-determining step seems different between each surface model. In the initial steps, because most of the H is located very near Cl atoms, the reaction rate is determined by the HCl evolution from Al–OH and Al–Cl. The reaction rate of HCl evolution at the surface (110)-(b) is significantly slower than in the other surface model, and this HCl reaction seems to be the rate-determining step. After the HCl evolution reaction occurred, the OH is seldom located next to Cl, and the rate-determining step is the H transfer between the O sites. This conclusion is quite similar to the previously reported theoretical studyAl2O3Firstprinciple ; CVDAl and it seems that our reactive MD simulation is reasonable for these three different surface models.
IV.4 Conclusion
A new method to optimize the reactive force field has been developed based on ML. Three important steps were introduced in this study: the -nearest neighbor algorithm to locate the possible local minima, a random forest regressor to construct the ML model, and grid search optimization to optimize the parameter set based on the ML model.
Using the ML technique, several optimized parameter sets that differ as far as possible could be obtained. All the obtained parameter sets reasonably reproduce the potential energy estimated by QM, but the physical properties are very different from each other, because the parameter optimization inevitably contains uncertainty for particular physical properties.
By evaluating the CN of Al and the O–Al–O angle, it is possible to know which parameter set is an appropriate choice for performing the MD simulation at high temperature. Then, as a pilot test, a reactive MD simulation was performed to analyze the HCl evolution reaction at the surface of -Al2O3. To this end, three different types of Cl–Al binding models were investigated, and the HCl evolution from Cl bridged between two Al atoms resulted in a significantly slower reaction rate than in other models. Such different types of Cl binding models could be reasonably evaluated by the parameter obtained in this study.
The proposed optimization process could simplify the complex process of ReaxFF parameter optimization with reasonable computer resources, which suggests that this strategy could be applied to simulate many reactive MD simulations in material science.
IV.5 ACKNOWLEDGMENTS
This research used the computational resources of the Supercomputer system ITO in R.I.I.T at Kyushu University as a national joint-usage/research center. We thank Professor Momoji Kubo of Institute for Materials Research, Tohoku University for the helpful discussions about the reactive MD simulation.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1(1) P. Deglmann, F. Furche, and R. Ahlrichs, Chem. Phys. Lett. 362 , 511 (2002).
- 2(2) Y. Alexeev, M. W. Schmidt, T. L. Windus, and M. S. Gordon, J. Comput. Chem. 28 , 1685 (2007).
- 3(3) A. Warshel and M. Karplus, J. Am. Chem. Soc. 94 , 5612 (1972).
- 4(4) J. A. Mc Cammon, B. R. Gelin, and M. Karplus, Nature 267 , 590 (1972).
- 5(5) S. Dapprich, I. Komáromi, K. S. Byun, K. Morokuma, and M. J. Frisch, J. Mol. Str.: THEOCHEM 461 , 1 (1999).
- 6(6) M. S. Gordon, D. G. Fedorov, S. R. Pruitt, and L. V. Slipchenko, Chem. Rev. 112 , 632 (2012).
- 7(7) P. Otto and J. Ladik, Chem. Phys. 8 , 192 (1975).
- 8(8) W. Yang, Phys. Rev. Lett. 66 , 1438 (1991).