Mixing thermodynamics and photocatalytic properties of GaP-ZnS solid solutions
Joel Shenoy, Judy N. Hart, Ricardo Grau-Crespo, Neil L. Allan, and, Claudio Cazorla

TL;DR
This study uses first-principles calculations to predict the stability and electronic properties of GaP-ZnS solid solutions, identifying compositions suitable for visible-light photocatalysis and guiding experimental efforts.
Contribution
It introduces a multi-configurational supercell approach to accurately predict mixing stability and electronic properties of GaP-ZnS solid solutions, aiding design of photocatalysts.
Findings
Zinc-blende phase is most energetically favorable.
Solid solution band gaps are within 2-3 eV range.
Certain compositions are metastable and promising for water splitting.
Abstract
Preparation of solid solutions represents an effective means to improve the photocatalytic properties of semiconductor-based materials. Nevertheless, the effects of site-occupancy disorder on the mixing stability and electronic properties of the resulting compounds are difficult to predict and consequently many experimental trials may be required before achieving enhanced photocatalytic activity. Here, we employ first-principles methods based on density functional theory to estimate the mixing free energy and the structural and electronic properties of (GaP)(ZnS) solid solutions, a representative semiconductor-based optoelectronic material. Our method relies on a multi-configurational supercell approach that takes into account the configurational and vibrational contributions to the free energy. Phase competition among the zinc-blende and wurtzite polymorphs is also…
Click any figure to enlarge with its caption.
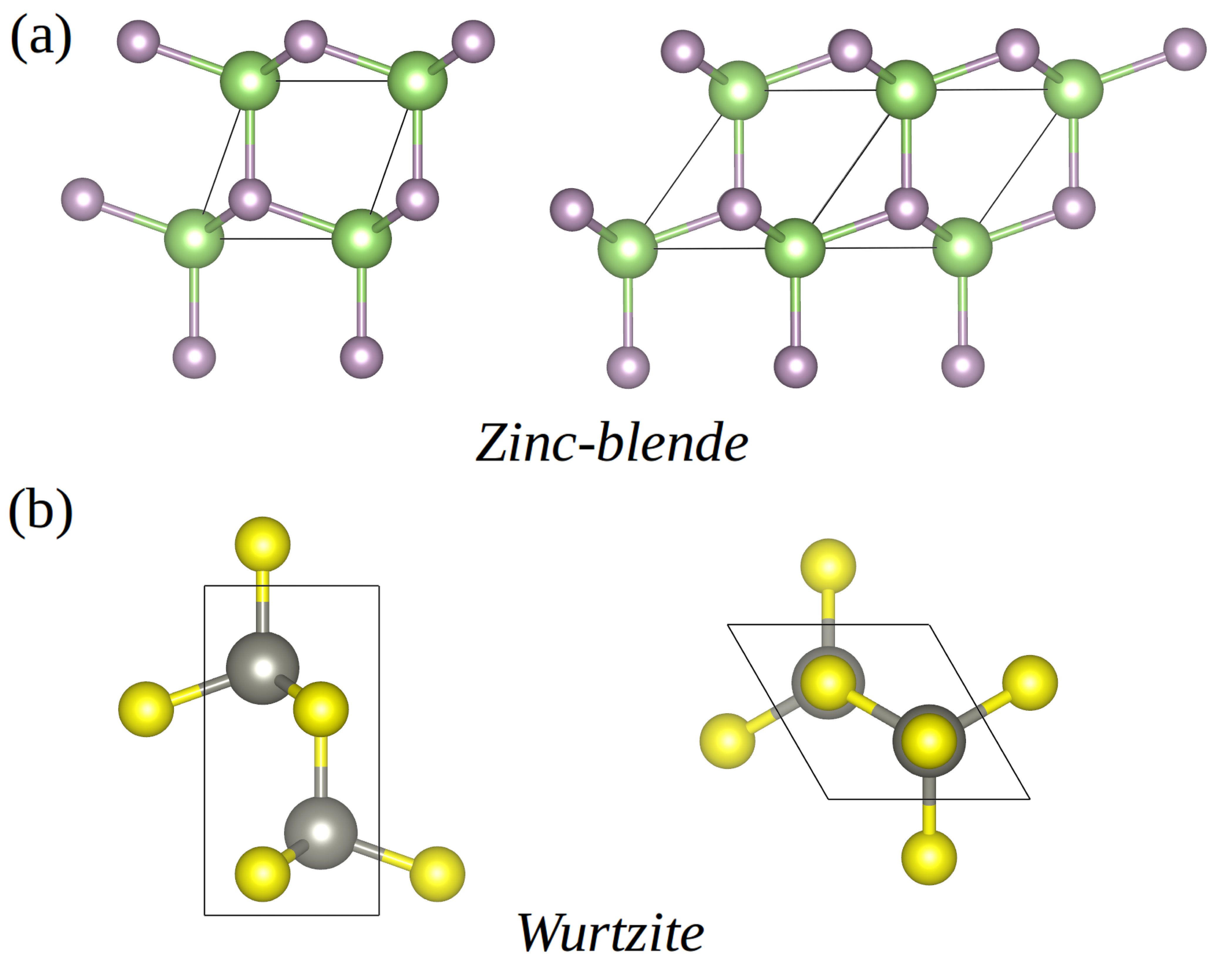 Figure 1
Figure 1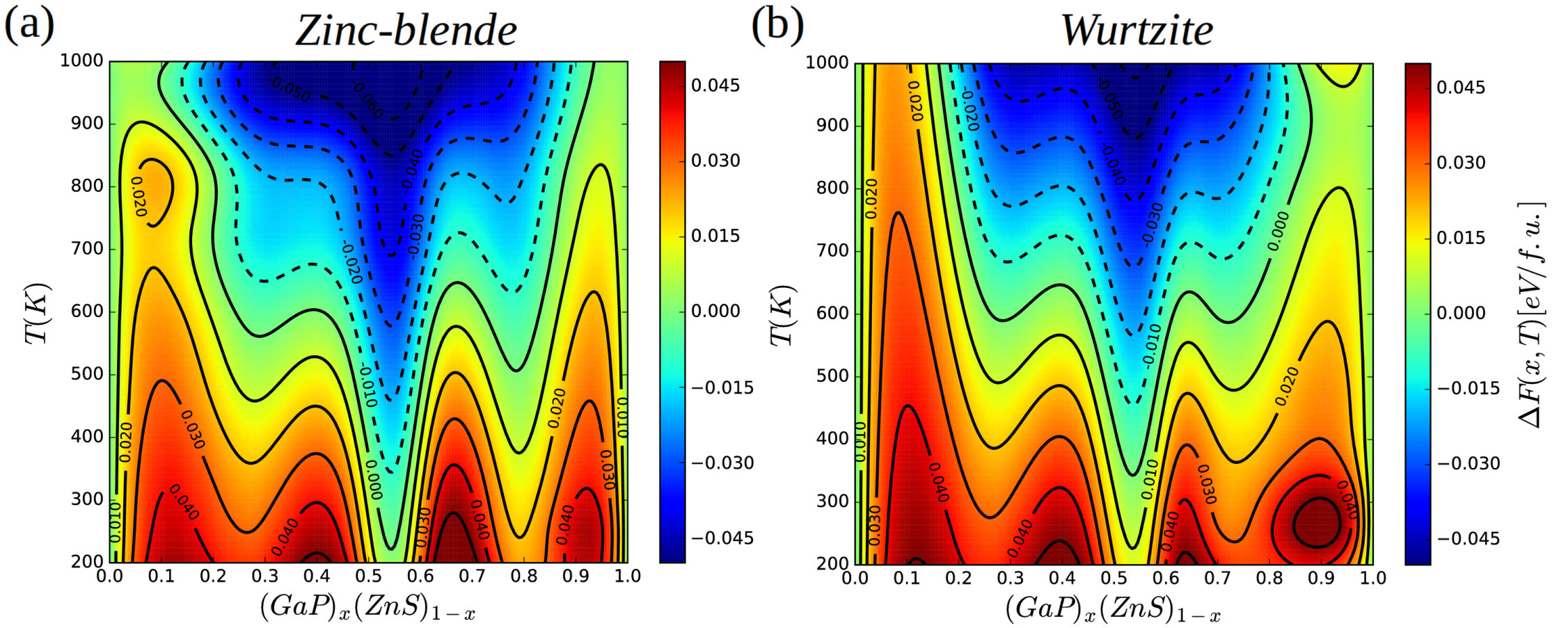 Figure 2
Figure 2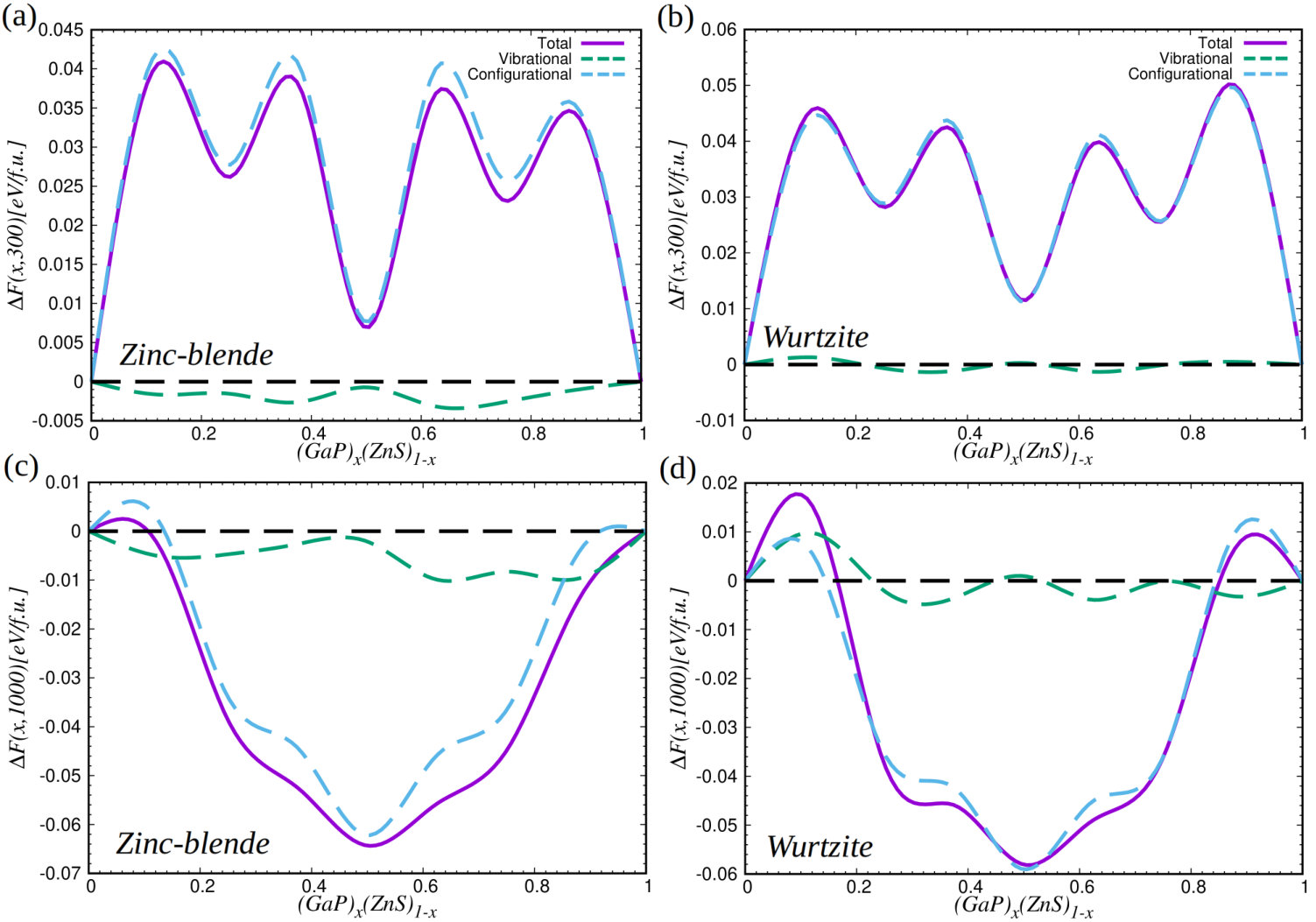 Figure 3
Figure 3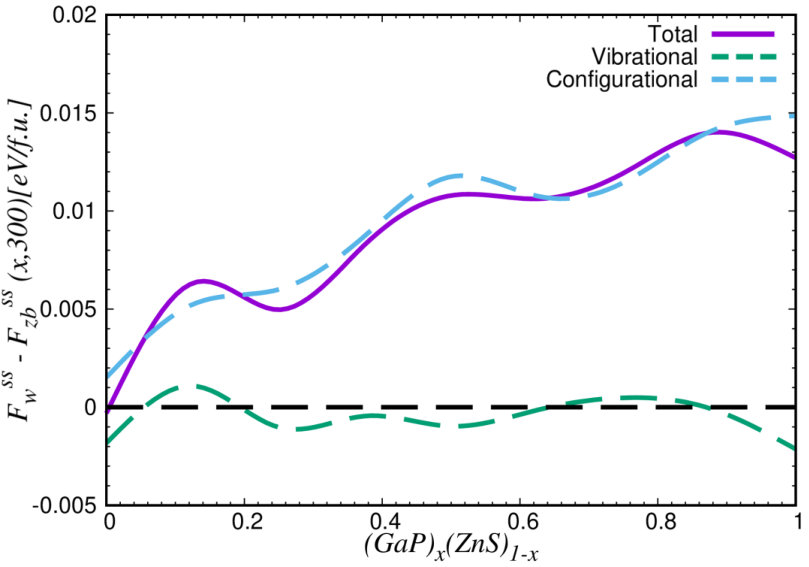 Figure 4
Figure 4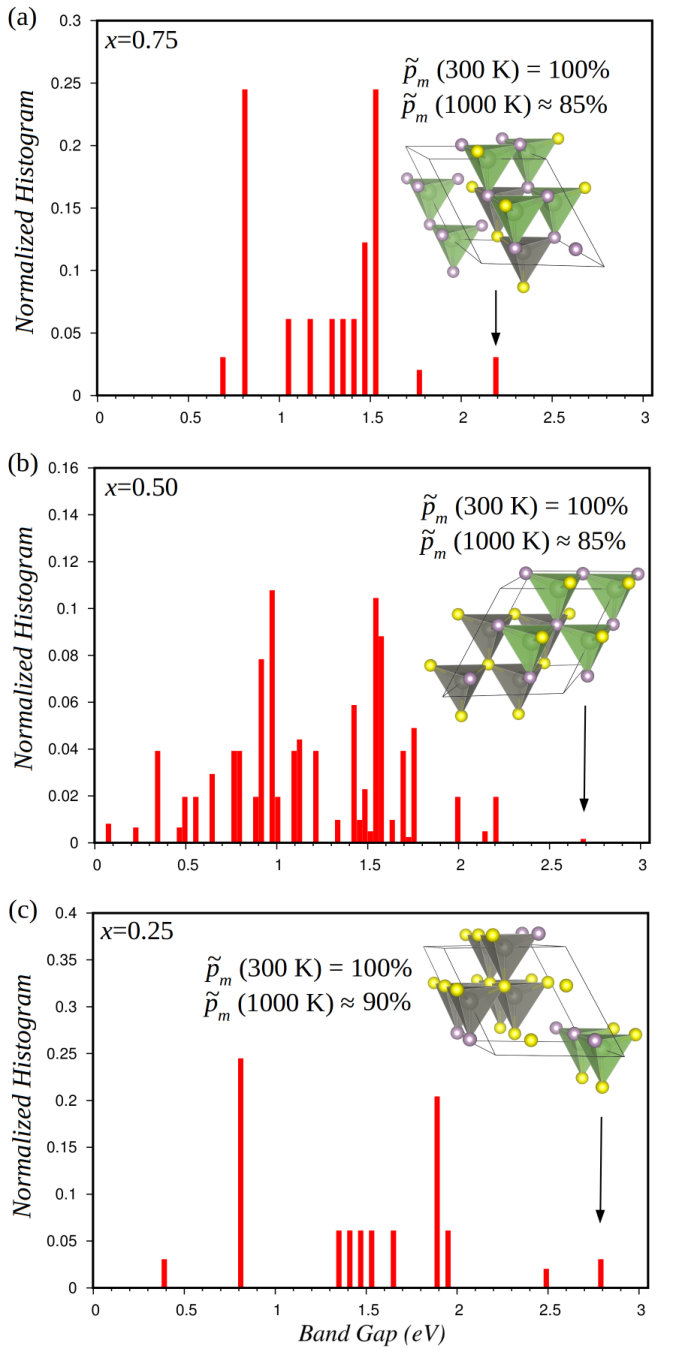 Figure 5
Figure 5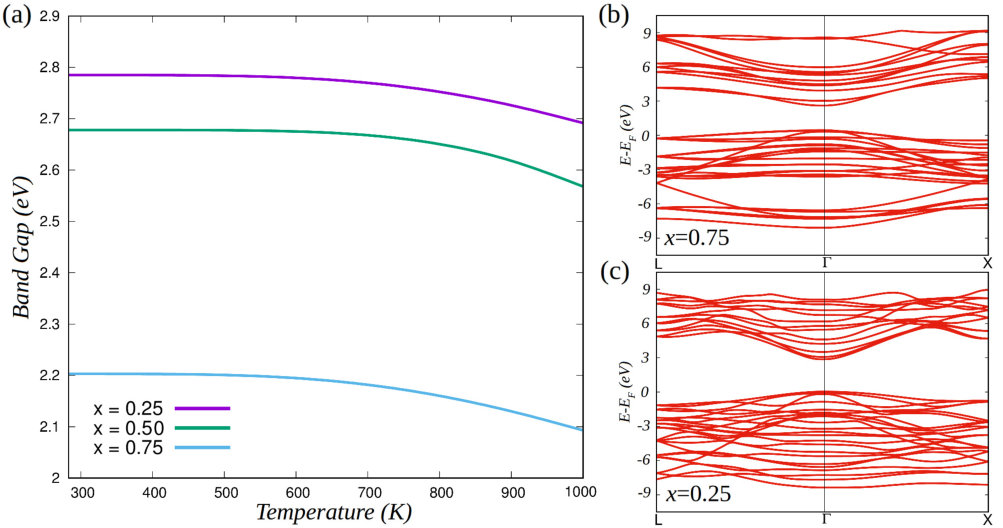 Figure 6
Figure 6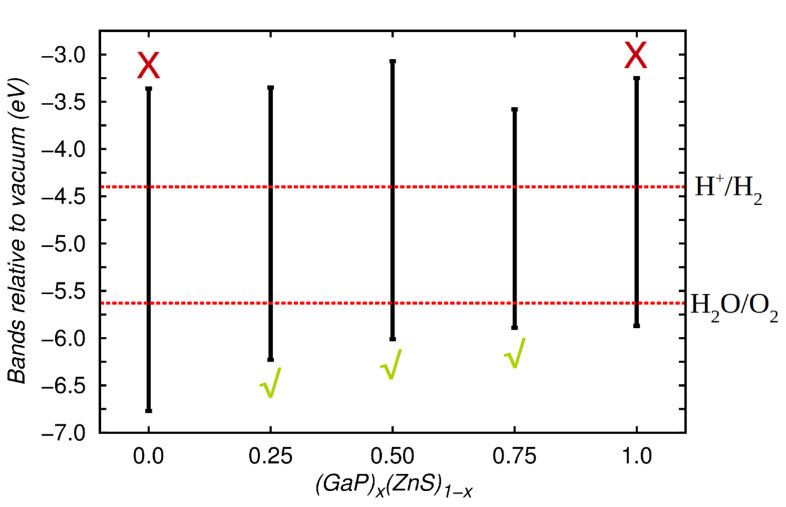 Figure 7
Figure 7Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Mixing thermodynamics and photocatalytic properties of GaP–ZnS solid solutions
Joel Shenoy1, Judy N. Hart1, Ricardo Grau-Crespo2, Neil L. Allan3, and Claudio Cazorla1
1School of Materials Science and Engineering, UNSW Sydney, NSW 2052, Australia
2Department of Chemistry, University of Reading, Reading RG6 6AD, United Kingdom
3School of Chemistry, University of Bristol, Cantock’s Close, Bristol BS8 1TS, United Kingdom
Abstract
Preparation of solid solutions represents an effective means to improve the photocatalytic properties of semiconductor-based materials. Nevertheless, the effects of site-occupancy disorder on the mixing stability and electronic properties of the resulting compounds are difficult to predict and consequently many experimental trials may be required before achieving enhanced photocatalytic activity. Here, we employ first-principles methods based on density functional theory to estimate the mixing free energy and the structural and electronic properties of (GaP)x(ZnS)1-x solid solutions, a representative semiconductor-based optoelectronic material. Our method relies on a multi-configurational supercell approach that takes into account the configurational and vibrational contributions to the free energy. Phase competition among the zinc-blende and wurtzite polymorphs is also considered. We demonstrate overall excellent agreement with the available experimental data: (1) zinc-blende emerges as the energetically most favorable phase, (2) the solid solution energy band gap lies within the – eV range for all compositions, and (3) the energy band gap of the solid solution is direct for compositions %. We find that at ambient conditions most (GaP)x(ZnS)1-x solid solutions are slightly unstable against decomposition into GaP- and ZnS-rich regions. Nevertheless, compositions , 50, and 75% render robust metastable states that owing to their favorable energy band gaps and band levels relative to vacuum are promising hydrogen evolution photocatalysts for water splitting under visible light. The employed theoretical approach provides valuable insights into the physicochemical properties of potential solid-solution photocatalysts and offers useful guides for their experimental realization.
I Introduction
Storing solar energy is critical for promoting the on–demand use of renewable energy sources. A promising solar-energy storage approach consists of generating hydrogen fuel by photocatalytic water splitting under sunlight kudo08 ; osterloh08 ; tong12 . For this scheme to progress it is necessary to find inexpensive and efficient photocatalytic materials. Binary semiconductors, such as TiO2, ZnS, and ZnO, have received great attention in this context owing to their natural abundance, structural simplicity, and scalable synthesis. While a direct band gap of around – eV is most desirable for photocatalytic applications kudo08 ; osterloh08 ; tong12 , binary semiconductors usually present wide and/or indirect energy band gaps that limit their absorption of visible light.
Solid solutions have emerged as an effective means to improve the photocatalytic performance of binary semiconductors. The main idea consists of mixing isostructural compounds with complementary electronic properties (e.g., a wide and direct energy band gap semiconductor with a narrow and indirect energy band gap semiconductor) in order to breed new materials with improved photocatalytic performance. Examples of solid-solution photocatalysts include (CdS)x(ZnS)1-x xing06 , (GaN)x(ZnO)1-x maeda06 , (LaCoO3)x(NaTaO3)1-x yi07 , and In1-xNixTaO4 zou01 .
Nevertheless, the effects of site-occupancy disorder, that is, non–periodic occupation of lattice sites in a crystal, on the electronic properties of solid solutions are challenging to foresee; consequently, many experimental attempts may be required before achieving any photocatalytic enhancement. In addition, inhomogeneity in the synthesized materials, caused by poor thermal stability of the compound mixtures, may limit performance. In this regard, computer simulations are useful since insightful analysis of the thermodynamic, structural, and electronic properties of photocatalytic materials can be performed in a systematic and cost-effective manner jensen08 ; valentin10 ; hart13 . Nevertheless, the non-periodic occupancy of lattice sites makes modelling of solid solutions challenging grau14 ; dieguez11 ; grau17 ; cazorla18 .
Several theoretical methods have been introduced in the literature to simulate solid solutions, which can be classified into three main categories. The first group involves approaches in which a sort of “average” atom is defined to recover the perfect periodicity of the system, which is beneficial from a computational point of view. In the context of first-principles calculations, this can be achieved via the virtual-crystal approximation, in which the potential felt by the electrons is obtained by averaging over multiple atoms bellaiche00 . In the second category of methods the effects of site-occupancy disorder are straightforwardly reproduced by randomly occupying the lattice sites in a large periodic supercell todorov04 ; allan01 . Since the involved supercell has to be large enough to mimic a random solution, this approach may be computationally very intensive. A special cell generation technique was introduced by Zunger et al. wei90 to construct quasi-random simulation supercells that are approachable with first-principles methods. The third type of solid-solution simulation method is typified by the multi-configurational supercell approach grau07 ; grau12 , in which the entire configurational space of a disordered system is first generated (for a finite-size supercell) and subsequently reduced to a managable set of inequivalent configurations by exploiting symmetry relations. Each inequivalent configuration is ascribed a Boltzmann-like occurrence probability that depends on its energy and configurational degeneracy, thus allowing for a full statistical treatment of the thermodynamic and functional properties of the material.
In this work, we present a comprehensive first-principles study of the mixing stability, and structural and electronic properties of (GaP)x(ZnS)1-x solid solutions at temperatures K, based on the multi-configurational supercell approach. GaP has an indirect energy band gap of eV while ZnS has a direct energy band gap at of eV, and previous computational studies have suggested that mixed GaP–ZnS compounds could be suitable for efficient absorption and emission of visible light hart13 . Indeed, (GaP)x(ZnS)1-x solid solutions have been experimentally studied in bulk shintani73 ; sonomura73 and as nanowires park14 , showing great promise as optoelectronic materials with band gap tunability and enhanced photoluminescence intensity. Bulk GaP and ZnS present two common polymorphs, zinc-blende and wurtzite (see Fig.1), and in the case of (GaP)x(ZnS)1-x nanowires a strong competition between the two phases has been reported park14 . For this reason we consider here both the zinc-blende and wurtzite polymorphs, which generally are observed in binary-octet semiconductors yeh92 . Our first-principles results show overall excellent agreement with the experimentally reported structural and electronic properties of GaP–ZnS solid solutions shintani73 ; sonomura73 . Interestingly, we predict that compositions , 50, and 75% render promising photocatalyst materials for production of hydrogen fuel from water splitting under visible light, able to meet the requirements of (1) a direct energy gap in the range of – eV, (2) a valence band level relative to vacuum lying below the water oxidation potential of eV, and (3) a conduction band level relative to vacuum lying above the hydrogen reduction potential of eV. The solid-solution simulation approach presented in this study, therefore, is very promising for the design and analysis of effective photocatalytic materials, which are pressingly needed for advancing the field of solar-energy storage.
II Computational Methods
II.1 Density functional theory calculations
We use the generalised gradient approximation to density functional theory (DFT) proposed by Perdew, Burke, and Ernzerhof (PBE) pbe96 as implemented in the VASP software package vasp . The projector augmented wave method is employed to represent the ionic cores bloch94 , considering the following electrons as valence states: Ga’s and ; P’s and ; Zn’s and ; and S’s and . Wave functions are represented in a plane-wave basis truncated at eV. For integrations within the Brillouin zone (BZ) we employ Monkhorst–Pack –point grids with a density equivalent to in the unit cell. By using these parameters we obtain zero-temperature energies converged to within meV per formula unit . Geometry relaxations are performed by using a conjugate–gradient algorithm that allows for cell volume and shape variations; the geometry relaxations are halted after the forces on the atoms fall below eVÅ*-1*. In order to reproduce site-occupancy disorder we adopt a –atom simulation cell constructed by replicating () times the elemental –atom (–atom) zinc-blende (wurtzite) unit cell (see next section).
We employ the hybrid HSE06 functional hse06 to estimate the electronic properties of equilibrium configurations previously generated with the PBE functional (see Supplementary Methods). This two-step approach of using PBE for generation of the configurational space, followed by HSE06 for electronic properties calculations, is required due to the high computational cost of hybrid functionals. The calculation of phonon frequencies is performed with the small displacement method kresse95 , in which the force-constant matrix is calculated in real-space by considering the proportionality between atomic displacements and forces alfe09 (see Supplementary Methods and Supplementary Fig.1). In order to estimate the positions relative to vacuum of the valence and conduction bands in (GaP)x(ZnS)1-x solid solutions we employ the CRYSTAL09 code crystal09 and the hybrid B3PW functional becke93 (see Supplementary Methods).
II.2 Multi-configurational supercell analysis
The mixing thermodynamics of a site disordered system with a constant number of atoms and at fixed temperature can be described as follows. For a complete set of possible system configurations (), a –dependent Boltzmann-like occurrence probability can be assigned to each:
[TABLE]
where is the corresponding energy, the Boltzmann constant, and the partition function defined as:
[TABLE]
Accordingly, the Helmholtz free energy of the system per formula unit (f.u.) can be estimated as:
[TABLE]
where is the number of formula units in the simulation cell (equal to the total number of atoms divided by two). The average value of the energy (or of any other well-defined quantity for each configuration ) in the corresponding configurational space adopts the form:
[TABLE]
In practice the configurational space of a chemically disordered solid may comprise a huge number of configurations (), hence normally it cannot be described with first-principles methods. An effective way to overcome such a limitation is to reduce the total number of possible configurations to by exploiting the symmetry properties of the parent crystal phase grau07 . In the generated reduced configurational space each inequivalent configuration has an associated degeneracy , equal to the number of symmetrically equivalent configurations with same energy , which all together fulfil the relation . The average value of the energy (or of any other well-defined quantity for each configuration ) in the reduced configurational space then is expressed as:
[TABLE]
where:
[TABLE]
The Helmholtz free-energy function in Eq.(3) accounts for configurational effects but neglects contributions from –induced lattice excitations, which a priori may be important in solid solutions walle02 . To include vibrational contributions in our free-energy calculations, we adopt the expression:
[TABLE]
where cazorla17 ; cazorla13 ; cazorla17b :
[TABLE]
In Eq.(8), represents the total number of wave vectors used for integration within the BZ, the vibrational eigenfrequencies of the system, and the summation runs over all wave vectors and phonon branches . Due to the huge computational expense associated with first-principles estimation of phonon excitations for a large number of configurations, we calculate just for the structure rendering the highest probability at room temperature (see Eq.6). We justify this choice in detail later when discussing our results; nevertheless, we estimate that the errors resulting from this procedure are within meV per formula unit (see Supplementary Methods).
To assess the mixing stability of bulk GaP–ZnS solid solutions, and since we consider zero-pressure conditions in this work, we estimate the corresponding mixing free-energy as a function of temperature and composition:
[TABLE]
where the superscripts indicate the system for which the Helmholtz free energy is calculated (“ss” stands for solid solution). A disordered system is thermodynamically stable against decomposition into GaP- and ZnS-rich regions if (although it may be unstable with respect to fluctuations in composition if ); otherwise, the system may be thermodynamically metastable or unstable (depending on whether is positive or negative). In this work, we perform the multi-configurational supercell calculations and accompanying statistical analysis with the SOD software grau07 . We use a –atom supercell that allows explicit simulation of (GaP)x(ZnS)1-x solid solutions at different compositions (namely, with ); results at other intermediate compositions are obtained via smooth spline interpolations. The resulting finite-size bias is estimated to be of the order of meV per formula unit (see tests performed for a larger supercell containing atoms explained in the Supplementary Methods).
Finally, we note that in the limit of very high temperatures the configurational entropy equals . Due to the finite size of the employed simulation cell, such a configurational entropy limit generally underestimates (in absolute value) the quantity:
[TABLE]
which is exact in the thermodynamic limit ( at constant composition ) and (the factor “2” in the formula above appears due to occupancy disorder in both the anion and cation sublattices). Aimed at correcting for such unavoidable finite-size bias, and as has been done in previous work grau12 ; becker00 , we apply the following shift to the configurational free energy:
[TABLE]
By doing this, the correct expression of the configurational free energy is consistently recovered in the limit. We note that, while this correction is quantitatively significant, the main conclusions presented in the following sections are not qualitatively affected by it (see Fig.2 and Supplementary Fig.2).
III Results and Discussion
III.1 Mixing thermodynamics
Figure 2 shows the mixing free-energy of GaP–ZnS solid solutions calculated as a function of structure, composition, and temperature at zero-pressure conditions (thermal expansion effects have been neglected). We have selected the temperature interval K because typical synthesis temperatures of semiconductor-based solid solutions are of the order of – K.
Below ambient conditions, we find that all zinc-blende bulk systems are thermodynamically unstable against decomposition into the end-members GaP and ZnS since , although some compositions may be kinetically stable where is locally positive (Fig.2a). However, at temperatures moderately above K some disordered crystals with compositions close to % become thermodynamically stable against decomposition into end-members GaP and ZnS, since . The composition range of this stability increases with increasing temperature, reaching % at 1000 K. Therefore, there exists the possibility of generating metastable solid solutions in that range of compositions at ambient temperature via quenching jones73 . The most favorable compositions for the realization of such metastable states are for close to , , and % since at a fixed temperature they render local minima in compositional space (hence is negative, so decomposition into GaP- and ZnS-rich regions can be kinetically hindered since small fluctuations in composition result in an increase in free energy). On the contrary, compositions near , , and % appear to be thermodynamically most unstable (since they render local maxima in compositional space, where is negative, and hence small fluctuations in composition result in a decrease in free energy).
The mixing properties of GaP–ZnS solid solutions in the wurtzite structure are very similar to those just described for the zinc-blende polymorph (Fig.2b). The same most favorable and most disadvantageous compositions for the synthesis of metastable systems are found, and at low temperatures the two corresponding maps are practically identical. However, as temperature is increased the mixing free-energy of the zinc-blende phase becomes slightly more favorable than that of the wurtzite polymorph. For instance, at K and % we estimate eV/f.u. in the wurtzite phase and eV/f.u. in the zinc-blende phase.
To understand the origins of the mixing stability differences between the zinc-blende and wurtzite polymorphs, we plot in Fig.3 the corresponding curves calculated at and K, split into different contributions (i.e., total, vibrational, and configurational). At room temperature (see Figs. 3a-b) the vibrational contribution to the mixing free-energy is practically negligible in both phases (see green lines therein), hence configurational effects are the dominant cause of the variations observed across the composition series. However, at high temperatures the vibrational mixing free-energy has a stabilizing effect in both phases, especially at compositions %, and is largest in absolute value in the zinc-blende polymorph (see Figs. 3c-d). For instance, at K and % amounts to meV/f.u. in the zinc-blende phase and to meV/f.u. in the wurtzite phase. The reason for such a vibrational mixing free-energy difference is that the high-frequency phonons, which are dominant at high temperatures, present lower energies in the zinc-blende phase (see Supplementary Fig.1). Meanwhile, configurational effects (blue lines in the figures) also tend to favor slightly the cubic polymorph over the hexagonal (e.g., at K and % meV/f.u. in the zinc-blende phase and meV/f.u. in the wurtzite phase).
Figure 4 shows the free-energy difference between GaP–ZnS solid solutions in the wurtzite and zinc-blende phases expressed as a function of composition at K. It is appreciated that the zinc-blende polymorph is energetically more favorable than the wurtzite phase at all compositions. Equivalent results are obtained also at higher temperatures (see Supplementary Fig.3). The main reason for the preference of the zinc-blende phase is that the configurational free energy is larger in absolute value than in the cubic polymorph. This is clearly appreciated in Fig.4, where we show that the vibrational free-energy differences between the two polymorphs are practically negligible as compared to the configurational counterparts (see blue and green lines therein). Hence our results indicate that the zinc-blende phase is more likely to be synthesized in practice than the wurtzite polymorph, which is consistent with the experimental observations shintani73 ; sonomura73 . In the remainder of the article, we will focus on zinc-blende solid solutions at compositions , , and % since these render the best thermodynamic stabilities.
III.2 Electronic properties
III.2.1 Energy band gap
In Fig.5, we show the normalized energy band gap histograms calculated with the HSE06 hybrid functional hse06 for zinc-blende solid solutions (Supplementary Methods). Specifically, is computed for each structure in the corresponding configurational ensemble and a normalized histogram is constructed subsequently by considering small energy increments (i.e., the number of structures within each specific band gap energy range is divided by the total number of structures –the occurrence probabilities are not considered here–). A large dispersion of energy band gap values spanning over the interval eV is observed. In the % case (Figs. 5b), we find particularly small values, that is, below eV, and the normalized histograms are quasi-continuous owing to the higher degree of site-occupancy disorder. It is worth noting that all the calculated energy band gaps are smaller than that of pure ZnS (i.e., eV heyd05 and eV) and lie within the range of visible or infrared light. Analogous results are obtained also for the wurtzite polymorph (see Supplementary Fig.4).
The large dispersions shown in Fig.5 indicate a strong dependence of the energy band gap, and in general of the electronic band structures of (GaP)x(ZnS)1-x solid solutions, on the local atomic environment. Similar behaviour is likely to occur in analogous binary octet semiconductors. For the zinc-blende (GaP)0.5(ZnS)0.5 system, we have performed a detailed analysis of the density of electronic states considering different atomic arrangements to better understand the origins of the variations. A clear electronic–structural correlation emerges from our calculations: the larger (smaller) the number of Zn–S bonds, or Ga–P bonds, the larger (smaller) the energy band gap. In turn, the electrostatic potential produced by local environments can be directly related to the number of Zn–S, or Ga–P, bonds. Specifically, when the number of Zn–S bonds, or Ga–P bonds, is minimized (leading to the smallest ) the electrostatic potential at the S and P atoms forming the top of the valence band (see Supplementary Fig.5) is lowest (i.e., their atomic environment is most negatively charged), hence the energy of the valence band is high. Likewise, the electrostatic potential at the Ga atoms forming the bottom of the conduction band (see Supplementary Fig.5) is highest when the number of Ga–P bonds, or Zn–S bonds, is minimized, which induces a lowering in the energy of the conduction band.
Remarkably, however, when the average value of is calculated with the multi-configurational supercell approach (see Eqs.5–6 in Sec. II.2), the band gap dispersions shown in Fig.5 play just a marginal role. As we explain next, this is due to the fact that one particular configuration is much more likely to occur than the rest (see the occurrence probabilities indicated in Fig.5 and the full spectra shown in Supplementary Fig.6).
Figure 6 shows the -dependent average value of the energy band gap calculated with the multi-configurational supercell method (see Eqs.(5)–(6)) for GaP–ZnS solid solutions at , , and %. presents an almost constant value throughout the whole temperature interval K at any composition, and increases from to eV as decreases from to %. As could have been foreseen, the energy band gap of the mixed compound is larger when the content of ZnS is larger (recall that and eV in pure GaP and ZnS hart13 ; heyd05 , respectively). The reason for the steady behaviour of as a function of temperature is that in all the analyzed cases the spectra of occurrence probabilities (see Eq. 6) very much favour the configuration with lowest energy (and low configurational degeneracy as shown by the histograms in Fig.5) over the others (Supplementary Fig.6). The lowest-energy configurations determined at each are sketched in Fig.5, along with their energy band gaps (see black arrows therein). As can be observed, the lowest-energy configurations render structures in which the number of Zn–S, or Ga–P, bonds are maximized, which as we have explained before correlates directly to largest energy band gaps.
The small variations observed in Fig.6a at K stem from the increasingly more important role that highly degenerate configurations (i.e., with large ’s –see Sec. II.2–) start to play at high temperatures (Supplementary Fig.6). Analogous energy band gap results are obtained also for the wurtzite phase (see Supplementary Fig.7). It is worth noting that the highly peaked nature of the distributions calculated in GaP–ZnS solid solutions (see Supplementary Fig.6) comes to justify the strategy that we have followed for estimating vibrational contributions to the mixing free-energy (see Sec. II.2).
The energy band gap results shown in Fig.6 are in very good agreement with the optical measurements performed by Shintani and Sonomura more than years ago shintani73 ; sonomura73 . In particular, the room-temperature experimental results –, –, and – eV obtained at , , and %, respectively, compare very well to our theoretical values , , and eV. Moreover, in our simulations we find that all compositions % render direct energy band gaps at (see Figs.6b,c, obtained for the most probable configurations), which also is consistent with the reported experimental obervations shintani73 ; sonomura73 . Thus, we confirm that zinc-blende (GaP)x(ZnS)1-x solid solutions display promising visible-light absorption features, namely, direct energy band gaps lying in the range – eV hart13 (likewise, the wurtzite polymorph possesses also promising electronic properties, see Supplementary Fig.7). Overall, our theoretical findings demonstrate that the present computational approach, which is based on the multi-configurational supercell method and density functional theory calculations, is able to reproduce closely the electronic band structure features of complex materials with site-occupancy disorder.
III.2.2 Energy band levels relative to vacuum
Promising photocatalytic materials should present not only suitable energy band gaps but also adequate energy band levels relative to vacuum moses11 . Here, we are interested in assessing the potential of (GaP)x(ZnS)1-x solid solutions for production of hydrogen fuel from water splitting under visible light. To this end, we calculated the position of the top of the valence band (VB) and bottom of the conduction band (CB) relative to the vacuum energy using the CRYSTAL09 code crystal09 with the methods described in Sec. II.1 and Supplementary Methods at compositions , , , , and %. In particular, we calculated the difference between the energy of the orbitals in bulk GaP and bulk ZnS and in the centre of ZnS and GaP slabs, . A non-polar –oriented slab with a total thickness equivalent to atomic layers was used for these calculations (corresponding to a thickness of Å for ZnS and of Å for GaP; such a length provides sufficiently well-converged results). The surface was selected because this has been reported to be the most stable for zinc-blende ZnS hamad02 ; wright98 and GaP hayashi82 . The cell length in the direction perpendicular to the slab surface was set to Å, giving a large enough vacuum gap to prevent interactions between periodic images of the slab. The slab was fully relaxed in all cases. The energy difference was then added to the VB and CB energies in order to obtain the energies relative to vacuum. For the solid solutions, the shift applied to the band energies was a weighted average of those for pure ZnS and pure GaP. This method has been previously applied to GaN–ZnO solid solutions valentin10 .
Figure 7 shows our hybrid B3PW becke93 results for the energy band levels relative to vacuum for different compositions. We reiterate that an ideal hydrogen photocatalyst material should present (1) a direct energy band gap lying in the range – eV, (2) a VB level relative to vacuum lying below the water oxidation potential of eV, and (3) a CB level relative to vacuum lying above the hydrogen reduction potential of eV. First, we note that our band alignment results obtained for bulk GaP and ZnS are in good agreement with previous first-principles calculations reported by other authors vandewalle03 . Pure GaP and ZnS are not suitable for photocatalytic water splitting since they do not fulfill condition (1) above. In contrast, all three solid solutions , , and % fulfill requirements (1)–(3) and hence are promising hydrogen photocatalyst materials for water splitting. For instance, in the % (%) case the VB level lies () eV below eV and the CB level is () eV above eV. We note that metastable ZnS-rich solid solutions, namely, cases and %, are of particular interest in terms of practical applications since GaP is scarce in nature and thus expensive, while ZnS is earth-abundant.
IV Summary
We have presented a comprehensive first-principles study of the mixing thermodynamics, and structural, electronic, and photocatalytic properties of (GaP)x(ZnS)1-x solid–solutions as a function of temperature and composition. Our theoretical approach relies on the multi-configurational supercell method, which allows a rigorous statistical treatment of site-occupancy disorder as well as calculation of accurate thermodynamic and functional properties of solid solutions. Valuable physical insights into the atomistic mechanisms behind the thermodynamic and functional features of solid solutions are attained on-the-go and interpreted easily in terms of probabilities (e.g., Boltzmann-like occurrence factors).
We find overall excellent agreement between our calculations and the experimental data reported on the structural and electronic properties of GaP–ZnS solid solutions. This good accordance demonstrates the accuracy and reliability of our employed computational method. Based on our enery band gap and band alignment results, we predict that compounds , , and % are very promising as photocatalyst materials for generation of hydrogen from water splitting under visible light. Similar theoretical studies to ours may be conducted for other encouraging combinations of semiconductor materials (e.g., GaN–ZnO and CdS–ZnS). Our theoretical approach is computationally affordable and fully general, hence it has the potential to accelerate the development of optimized materials based on semiconductor solid solutions for solar-energy storage; also, to improve the design of materials for a range of other applications in which tuning of the opto-electronic behavior is important. We envisage that our approach will promote and assist the experimental synthesis of bettered photocatalysts by providing useful insights into their mixing thermodynamics.
Acknowledgements.
This research was supported under the Australian Research Council’s Future Fellowship funding scheme (No. FT140100135). Computational resources and technical assistance were provided by the Australian Government and the Government of Western Australia through the National Computational Infrastructure (NCI) and Magnus under the National Computational Merit Allocation Scheme and The Pawsey Supercomputing Centre.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1(1) A. Kudo, Y. Miseki, Chem. Soc. Rev. 2009 , 38 , 253.
- 2(2) F. E. Osterloh, Chem. Mater. 2008 , 20 , 35.
- 3(3) H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri, J. Ye, Adv. Mater. 2012 , 24 , 229.
- 4(4) C. Xing, Y. Zhang, W. Yan, L. Guo, Int. J. Hydrog. Energy 2006 , 31 , 2018.
- 5(5) K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue, K. Domen, Nature 2006 , 440 , 295.
- 6(6) Z. G. Yi, J. H. Ye, Appl. Phys. Lett. 2007 , 91 , 254108.
- 7(7) Z. Zou, J. Ye, K. Sayama, H. Arakawa, Nature 2001 , 414 , 625.
- 8(8) L. L. Jensen, J. T. Muckerman, M. D. Newton, J. Phys. Chem. C 2008 , 112 , 3439.