Adsorption and dissociation of iron phthalocyanine on H/Si(111): Impact of van-der-Waals interactions and perspectives for subsurface doping
Benjamin Geisler, Peter Kratzer

TL;DR
This study investigates the adsorption behavior of iron phthalocyanine on H/Si(111), emphasizing van-der-Waals interactions, and explores implications for subsurface doping with detailed first-principles calculations and STM simulations.
Contribution
It provides a comparative analysis of van-der-Waals interaction approaches and discusses potential subsurface doping reactions of FePc on H/Si(111).
Findings
FePc is mainly physisorbed with a 2.6 Å distance from the surface.
Simulated STM images match experimental data well.
Fe is strongly bound in the molecule with about 9.6 eV binding energy.
Abstract
The adsorption of iron phthalocyanine (FePc) on the passivated H/Si(111) surface is explored from first principles. We find that the organic molecule is predominantly physisorbed with a distance to the surface of Angstrom, but also exhibits sizable resonance with the underlying substrate. This establishes the present system as interesting mixed covalent-van-der-Waals-bound test case, which we use to compare the impact of different approaches to van-der-Waals interactions. (Spin-polarized) scanning tunneling microscopy (SP STM) images are simulated, selectively accessing different molecular orbitals via the applied bias voltage in the spirit of scanning tunneling spectroscopy. Comparison with experimental STM images reveals very good agreement. We find a significant magnetic contrast exceeding Angstrom in the SP STM images for and V. Binding energies of…
Click any figure to enlarge with its caption.
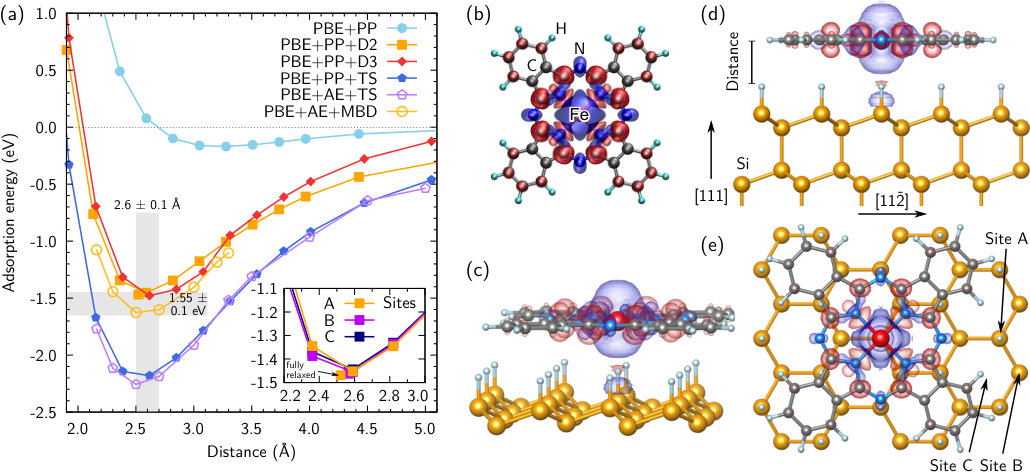 Figure 1
Figure 1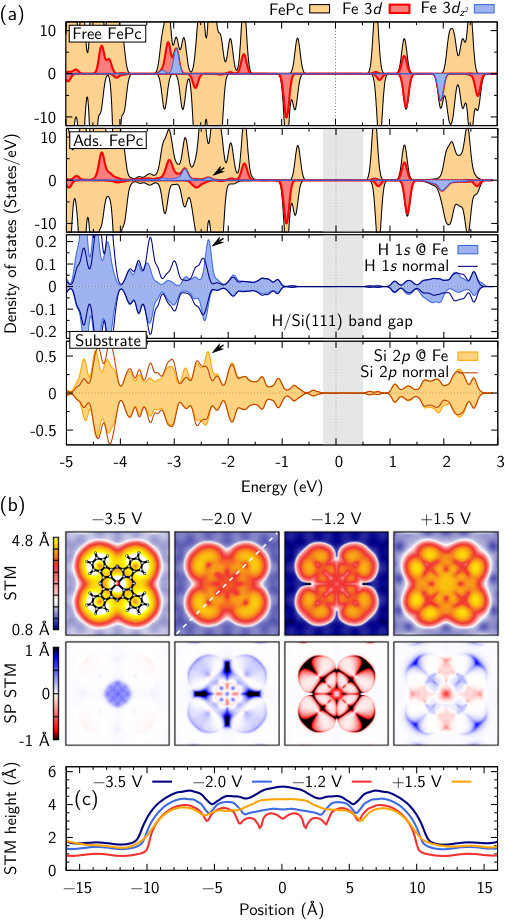 Figure 2
Figure 2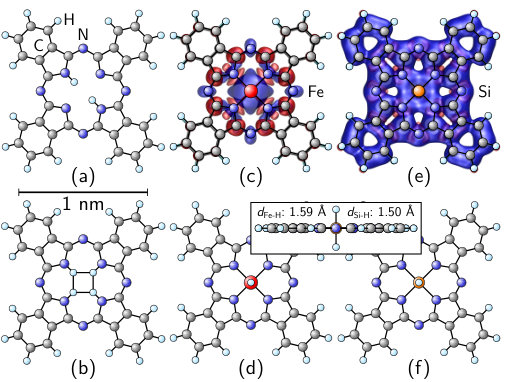 Figure 3
Figure 3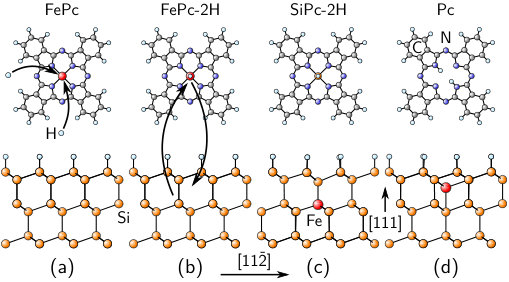 Figure 4
Figure 4| PP | AE | |||||
|---|---|---|---|---|---|---|
| PBE | D2 | D3 | TS | TS | MBD | |
| Distance (Å) | ||||||
| (eV) | ||||||
| Fe | Fe 2H | Si | Si 2H | 2H | 4H | |
|---|---|---|---|---|---|---|
| (eV) | ||||||
| () |
| PP | AE | ||||||
|---|---|---|---|---|---|---|---|
| PBE | PBE | LDA | B3LYP | PBE0 | HSE06 | HF | |
| Fe in FePc | |||||||
| 2H in Pc | |||||||
| Reaction | (eV) |
|---|---|
| FePc pure H/Si | |
| Fe subs. SiPc | |
| Fe subs. SiPc-2H H-Si bond broken | |
| Fe subs. complex with Si self-inter. empty Pc ring | |
| Fe subs. Si self-inter. empty Pc ring | |
| Fe inter. empty Pc ring | |
| Fe inter. Pc H-Si bond broken | |
| FePc-2H pure H/Si | |
| Fe subs. SiPc-2H | |
| Fe subs. complex with Si self-inter. Pc | |
| Fe inter. Pc |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Adsorption and dissociation of iron phthalocyanine on H/Si:
Impact of van-der-Waals interactions and perspectives for subsurface doping
Benjamin Geisler
Fakultät für Physik, Universität Duisburg-Essen and Center for Nanointegration (CENIDE), Campus Duisburg, Lotharstr. 1, 47048 Duisburg, Germany
Peter Kratzer
Fakultät für Physik, Universität Duisburg-Essen and Center for Nanointegration (CENIDE), Campus Duisburg, Lotharstr. 1, 47048 Duisburg, Germany
(March 17, 2024)
Abstract
The adsorption of iron phthalocyanine (FePc) on the passivated H/Si surface is explored from first principles. We find that the organic molecule is predominantly physisorbed with a distance to the surface of and an adsorption energy of eV, but also exhibits sizable resonance with the underlying substrate. This establishes the present system as interesting mixed covalent-van-der-Waals-bound test case, which we use to compare the impact of different approaches to van-der-Waals interactions. (Spin-polarized) scanning tunneling microscopy (SP STM) images are simulated, selectively accessing different molecular orbitals via the applied bias voltage in the spirit of scanning tunneling spectroscopy. Comparison with experimental STM images reveals very good agreement. We report a significant magnetic contrast exceeding in the SP STM images for and V. Aiming for a magnetic functionalization of Si for possible spintronics applications, magnetic moments and binding energies of different (transition metal) atoms in the center of the Pc ring are presented, which particularly show that Fe is strongly bound in the molecule (about eV). Finally, we discuss different mechanisms for subsurface Fe doping by room-temperature FePc deposition and point out two feasible reactions. Concomitantly, we identify the crucial role of a preceding destabilization of FePc, for instance, by pre-adsorbed H atoms, which subsequently strongly stabilize the final state of the reaction.
I Introduction
The exploration of viable routes towards Si-based spintronics devices Jansen (2012) is of key importance, owing to the abundance of Si and its unmatched role in modern semiconductor technology. Magnetic functionalization of Si by a controlled doping with transition metal (TM) impurities is a potential strategy. Despite intense research, comprising recent studies of formation energies and magnetic properties of isolated TM impurities and impurity pairs Zhang et al. (2008), the dependence of magnetism on the doping concentration Küwen et al. (2009); Shaughnessy et al. (2010), or Mn doping of Si Qian et al. (2006); Wu et al. (2007); Otrokov et al. (2011), at present Mn-doped GaAs dominates the field Ohno et al. (1996); Koenraad and Flatté (2011); Jancu et al. (2008).
For a rational materials design, it is crucial to improve the fundamental understanding of the interaction mechanisms between impurities and the surrounding host matrix. We recently promoted (spin-polarized) scanning tunneling microscopy (SP STM) Wiesendanger (2009) as a powerful method to explore bulklike interaction properties of impurities in semiconductors on the atomic scale by exploiting passivated surfaces Geisler and Kratzer (2015). Specifically, we focused on TM impurities below the H/Si surface. The passivating H layer could be added after growth and subsequent cleavage of a sample; however, exposing the doped system to a wet-chemical treatment Gruyters et al. (2013) will cause strong reactions of the TM impurities with H. Hence, a strategy to achieve a controlled subsurface doping of the H/Si system after preparation would be valuable. It was claimed recently that this can be accomplished by the adsorption of iron phthalocyanine (FePc) on the H/Si surface Gruyters et al. (2013). This is particularly compelling owing to the fact that Fe is one of the fundamentally most interesting impurities in Si, since its magnetic moment could not be unambiguously determined from first principles so far Geisler and Kratzer (2015).
The study of surface reactions in hybrid inorganic-organic systems is a rich topic of its own. Considerable research focuses on the adsorption of molecules on metallic Morbec and Kratzer (2017); Herper et al. (2014); Ruiz et al. (2012), semiconducting Veiga et al. (2016); Gruyters et al. (2012); Sena et al. (2009), or insulating Repp et al. (2005) surfaces, magnetic properties of TM-based molecules and their interactions with ferro- and nonmagnetic substrates Bernien et al. (2009); Atodiresei et al. (2010), and magnetic switching phenomena Bhandary et al. (2011), which aim at spin-dependent molecular electronics Wende et al. (2007); Kratzer et al. (2017). The Kondo effect can be observed in magnetic molecules deposited on metal surfaces Fu et al. (2007); Perera et al. (2010). Given that the impact of the surface on the electronic structure of the adsorbate is minute, these systems are also attractive models to gain fundamental insight by comparing experimental and time-dependent density functional theory (TDDFT) results, for instance, on optical properties of Pc-derived molecules Cocchi et al. (2014). Naturally, van-der-Waals (vdW) interactions are of central relevance in this field.
Here we explore the adsorption of FePc on H/Si from first principles. We find that the organic molecule is predominantly physisorbed, but also shows significant resonance with the underlying substrate. We use this paradigmatic case of a system in a mixed covalent-vdW-bonding state to compare the impact of different established Grimme (2006); Grimme et al. (2010) and recent Tkatchenko and Scheffler (2009); Tkatchenko et al. (2012) approaches to vdW interactions, thereby going beyond small model systems that are conventionally used for optimizing and testing these dispersion methods. Analogies to 2D materials, e.g., graphene, boron nitride, or transition metal dichalcogenides add to the relevance of such benchmarking Tawfik et al. (2018). (SP) STM images are simulated, selectively accessing different molecular orbitals via the applied bias voltage in the spirit of scanning tunneling spectroscopy, and compared to experiment. The SP STM images exhibit a magnetic contrast that exceeds for and V. In order to shed light on the mechanism behind the suggested subsurface doping involving room-temperature FePc deposition, we present binding energies of different (TM) atoms in the center of the Pc ring, which particularly reveal that Fe is strongly bound (about eV). This result is confirmed by different methodologies, including hybrid functionals, and holds for other TM centers as well. Despite this surprising finding, we identify two feasible reactions by analyzing several distinct mechanisms to achieve subsurface Fe doping. Simultaneously, we highlight the crucial role of a preceding destabilization of the reactant FePc, for instance, by pre-adsorbed H atoms, which subsequently strongly stabilize the final state of the reaction.
II Methodology
We performed spin-polarized density functional theory Kohn and Sham (1965) (DFT) calculations within the plane-wave and ultrasoft pseudopotential (PP) framework as implemented in the Quantum Espresso code Vanderbilt (1990); Giannozzi et al. (2009) and compare to results obtained with the full-potential, all-electron (AE) FHI-aims code Blum et al. (2009) which employs numeric atom-centered basis functions. We use cutoff energies for wave functions and density of and Ry and a ’tight’ ’tier2’ basis, respectively. Gas-phase molecules were simulated with different exchange-correlation functionals, namely semilocal LDA Ceperley and Alder (1980); Perdew and Wang (1992), PBE Perdew et al. (1996a), the hybrid functionals HSE06 Heyd et al. (2006), B3LYP Becke (1993); Lee et al. (1988); Stephens et al. (1994), PBE0 Perdew et al. (1996b), and in the Hartree-Fock approximation. For the adsorption and STM simulations, we used a Si- slab at the PBE lattice constant of Si () with two passivating H layers, vacuum region, a -point grid including the point and the PBE functional for exchange and correlation. Van-der-Waals interactions were treated within the Grimme D2 Grimme (2006); Barone et al. (2009) and D3 Grimme et al. (2010) approaches and compared to the Tkatchenko-Scheffler (TS) method Tkatchenko and Scheffler (2009), in which the free-atom coefficients, polarizabilities, and vdW radii are rescaled by a Hirshfeld partitioning of the electron density. We also explored the more recent many-body dispersion (MBD) technique Tkatchenko et al. (2012), in which the atomic response functions are represented by quantum harmonic oscillators and the screened long-range many-body vdW energy is computed from the adiabatic connection fluctuation dissipation theorem within the dipole approximation. Constant-current STM images at a bias voltage were simulated subsequently in the spirit of Tersoff and Hamann Tersoff and Hamann (1983) as -isosurfaces of the integrated local density of states (ILDOS) Geisler et al. (2012); Suzuki et al. (2013); Geisler and Kratzer (2013, 2015); Geisler and Pentcheva (2019): , ; we used . SP STM images correspond to differences of two STM images derived from the individual spin channels.
The DFT treatment of FePc is nontrivial due to numerical instabilities. Calculations employing the PBE exchange-correlation functional often lead to a symmetry-broken ground state, whereas hybrid functionals such as B3LYP preserve the symmetry of the molecule Marom and Kronik (2009). We used the B3LYP electronic structure as reference, which we found to be qualitatively reproduced by PBE Cococcioni and de Gironcoli (2005) with a small eV, as is has also been utilized in the literature Mugarza et al. (2012); Herper et al. (2014). In order to obtain unbiased binding energies, we did not apply a Hubbard in those cases, assuring carefully that the symmetry is not broken.
III Adsorption of FePc on H/Si
Figure 1(a) shows the adsorption energies of a single FePc molecule on the H/Si surface as functions of the molecule-surface distance calculated from DFT total energies:
[TABLE]
The adsorption energy curves exhibit a typical potential shape, diverging for small distances and approaching zero (corresponding to the total energy sum of separated systems) from below for large distances. Three distinct adsorption sites are compared: centering Fe above the first Si layer positions, i.e., above one of the H atoms (A), above the second/third Si layer positions (B), and above the fourth Si layer positions (C), which correspond to the hollow centers of the topmost bilayer rings [Fig. 1(e)]. The curves have been obtained from rigid atomic structures, i.e., no relaxation beyond the individual equilibrium geometries of molecule and surface has been performed. Subsequent calculations optimizing all atomic positions at the adsorption distance have shown that adsorption at site A is most stable [Fig. 1(a)]. Energy differences with respect to sites B and C amount to only a few meV; the same applies to different rotation angles of the molecule. Thus, we conclude that FePc is highly mobile on the H/Si surface.
The resulting adsorption energies and distances are summarized in Table 1. Interestingly, already PBE without further vdW corrections provides a shallow adsorption minimum of eV near despite the chemical inertness Neuwald et al. (1992) of the H/Si surface. However, this adsorption energy is certainly far too low, and the equilibrium molecule-surface distance will be severely overestimated. Indeed, we find that FePc attaches more closely to the substrate if vdW interactions are taken into account; all considered vdW methods provide adsorption distances within an interval of . The adsorption energies are more strongly spread: Predictions based on the D2, D3, and MBD approaches range from to eV, whereas the TS method results in a much stronger binding of about to eV. Thus, a many-body treatment of the vdW interactions Tkatchenko et al. (2012) corroborates the results of D2 (and D3). Interestingly, we find very good agreement for two distinct implementations (PP vs. AE) of the TS method in two individual codes [Fig. 1(a)].
The results shown in Fig. 1(a) and Table 1 can be compared to other hybrid inorganic-organic systems. For 3,4,9,10-perylene tetracarboxylic dianhydride (PTCDA) on Ag Ruiz et al. (2012), PBE provides a shallow adsorption minimum of about eV at a distance larger than the experimental value. Inclusion of D2 vdW corrections severely overestimates the adsorption energy by eV (), simultaneously providing an almost correct molecule-surface distance. In that case, screening effects of the metal substrate play an important role, and the performance of D2 will be significantly better for the present system. For anthracene and pentacene on Ag Morbec and Kratzer (2017), PBE predicts a shallow adsorption minimum of eV, and the surface-molecule distance is lowered by about after inclusion of vdW interactions. The adsorption energy obtained with the MBD approach is in almost perfect agreement with the experimental value.
For the related case of vdW-bound 2D materials, recent comparative work revealed that the TS method predicts the adsorption energy with least precision, whereas D2, D3, and in particular a many-body treatment of the vdW interactions considerably improve the accuracy Tawfik et al. (2018). In conjunction with the analysis above, we therefore conclude a tentative adsorption energy of eV for FePc on H/Si.
The environment of the Fe2+ ion results in a FePc magnetic moment of as expected from ligand field theory. Its spin density is shown in Fig. 1(b). The adsorbed molecule retains its gas-phase magnetic moment for all methodologies considered here, and it becomes obvious from Figs. 1(c)-(e) that also the shape of the spin density is largely preserved, apart from a slight compression of the lobe directed towards the substrate. Interestingly, we observe a sizable spin polarization of the H-Si bond in the substrate below the Fe ion.
At first sight, it appears that the proximity of the surface has only modest impact on the electronic structure of the molecule [Fig. 2(a)], which is different e.g. for anthracene and pentacene on Ag Morbec and Kratzer (2017). Relative to gas-phase FePc, the band gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is almost unaffected. Both reside within the valence band or the conduction band of the substrate, respectively. A finite density of states emerges at eV. Specifically, the Fe states are strongly broadened; while the occupied majority-spin state at eV shifts to eV, its unoccupied equivalent in the minority-spin channel retains its position. The H and Si states below the Fe ion clearly respond to the presence of the molecule as well, reflecting the induced spin polarization and exhibiting an Fe--H--Si- resonance [Fig. 2(a)]. Close inspection of the atomic positions reveals that Fe and H leave their respective planes and reduce their distance by . This attractive interaction shows that the system enters a mixed covalent-vdW-bonding state, which exemplifies that the inclusion of vdW interactions does not only quantitatively improve the results, but is of qualitative importance for a proper modeling of FePc adsorption on H/Si. Hence, this system emerges as interesting test case for different vdW corrections in the field of electronic structure theory Grimme (2006); Grimme et al. (2010); Tkatchenko and Scheffler (2009); Tkatchenko et al. (2012). Specifically, exploring the performance of ’true’ vdW density functionals is of importance. Dion et al. (2004); Román-Pérez and Soler (2009); Lee et al. (2010).
Figure 2(b) shows simulated STM and SP STM images for an adsorbed single FePc molecule on the H/Si surface at site A, together with a line scan for different bias voltages [Fig. 2(c)]. For the filled-state images (), the H sites of the substrate can be identified by white spots Geisler and Kratzer (2015). The symmetry of the molecule is clearly reflected in all panels. The distinct lobes that can be seen for V (cross-like shape) merge for voltages of higher magnitude (flower-like shape), consistent with experiment Gruyters et al. (2012). Moreover, while for V the outer parts of the lobes are dominant, a feature appears in the center of the molecule with increasing bias voltage that can be clearly seen from the line scans and is also observed experimentally Gruyters et al. (2012). In our simulations, the molecule is detected - above its physical position and - above the substrate-related background, with an apparent diameter of (structurally ). Experimentally, its signature appears a little larger, exhibiting a height of above the substrate-related background and a diameter of at to V and - pA Gruyters et al. (2012). Overall, the agreement between simulations and experiment is very good.
We find that the SP STM magnetic contrast is significant, exceeding for and V. Since SP STM does not correspond to a mapping of the spin density (Fig. 1), but of the states near the Fermi energy, we see a negative response for V (red, largest at the outer lobe parts) that switches to a positive signal for V (blue, largest between the lobes). Finally, inclusion of the majority-spin Fe state for a bias voltage of V leads to a positive feature in the center. In turn, this feature appears negative and even more pronounced for a mapping of the unoccupied states beyond V (not shown).
IV Binding energies of transition metal ions in Pc molecules
Experiments have suggested that FePc molecules can release their Fe center upon room-temperature adsorption on H/Si, thereby leading to a subsurface doping of H/Si by Fe Gruyters et al. (2013). In order to shed light on the underlying mechanism, it is important to understand the energetics of TM bonding in the Pc ring. To give additional credibility to our results, we carried out calculations for various gas-phase Pc molecules not only with Fe, but also with other TM atoms and with Si as center, and compare the values obtained with a variety of exchange-correlation functionals. This information will further be used in the next section to extract DFT total energy differences of distinct reactions at the H/Si surface.
Figure 3 shows optimized atomic structures of selected Pc molecules. The corresponding binding energies of different central atoms in an empty Pc ring are given in Table 2. They are defined as
[TABLE]
where the first two terms are the total energies of the molecule and the empty Pc ring as reference, is the difference of atoms of species between molecule and reference, and denotes the total energy of an isolated atom of species . Two H atoms are bound twice as strong in the Pc ring ( eV) as in a H2 molecule (PBE: eV), while four bound H atoms are less favorable than the former case plus an isolated H2 molecule. Most notably, we find that Fe is even more strongly bound ( eV). In order to see if this surprisingly high Fe binding energy is an exception, we also studied other TM centers in the Pc ring and found similar results, CuTi exhibiting the lowesthighest binding energy ( eV). To account for possible Si exchange processes between substrate and molecule, we also simulated a Si atom in a Pc ring, and found it to be bound with eV, a value almost twice as high as in Si bulk (PBE: eV). Both FePc and SiPc can bind two additional H atoms above and below the molecular plane. While in the case of FePc the formation of an isolated H2 molecule is energetically favored, SiPc-2H is stable against decomposition with an energy gain of eV.
In addition to the binding energies, Table 2 also lists the spin magnetic moments of different molecules. The values for the more common types such as MnPc, FePc, CoPc, or CuPc are well known Fu et al. (2007); Mugarza et al. (2012). We also find (high) magnetic moments for TiPc and, particularly, CrPc. Most peculiarly, we observe a very stable magnetic moment of for SiPc, the spin density extending over the entire molecule [Fig. 3(e)], while that of FePc is confined to the center.
In order to further validate the very high FePc binding energy we reported above, Table 3 compares results calculated with different methodologies (PP vs. AE) and exchange-correlation functionals for FePc and a Pc ring with two H atoms [Fig. 3(a)]. In particular, hybrid functionals like B3LYP, PBE0, or HSE06 are commonly used for organic molecules Åhlund et al. (2006); Heyd et al. (2003); Krukau et al. (2006). The PBE results obtained within the PP and AE frameworks are in excellent agreement. LDA shows the well-known overbinding. For FePc, the binding energies provided by the hybrid functionals are roughly eV smaller than the PBE results, but still very high. For the Pc ring with two H atoms, hybrid functional results are similar to PBE. Hartree-Fock calculations lead to significantly smaller binding energies for both systems, but since they ignore all correlation effects, their appropriateness for larger organic molecules can be questioned. The magnetic moment of FePc is in all cases. While Fe is more strongly bound than two H atoms in LDA and PBE, the reversed prediction is made by hybrid functionals and Hartree-Fock. HSE06 and PBE0 provide very similar results, which shows that the screening of the Coulomb interaction in HSE06 is irrelevant on the present length scale.
On the basis of these first-principles findings we conclude that the very high binding energy we obtained for a Fe center in a Pc ring is indeed reasonable.
V Subsurface doping
After this excursion, we now return to FePc and possible mechanisms to achieve subsurface Fe doping by room-temperature deposition of FePc on H/Si, as it has been proposed recently on the basis of STM imaging Gruyters et al. (2013). The comprehensive DFT studies presented in the previous section, which are summarized in Tables 2 and 3, show that Fe is very strongly bound to the organic part of the molecule and thus quite difficult to extract. This raises the question whether the suggested doping strategy is realistic, particularly since a combined experimental-theoretical analysis of the STM observations remained inconclusive Gruyters et al. (2013); Geisler and Kratzer (2015).
In a first step towards addressing this complex question, DFT total energy differences of several distinct reactions (Table 4) can be considered under the constraint that the number of atoms of each species has to be conserved. Positive indicates an endothermic reaction. For instance, the Fe atom could leave the FePc molecule and replace a Si atom in the host (substitutional Fe); subsequently, the Si atom is incorporated in the rest of the molecule, forming SiPc in which the Si atom is more strongly bound than in bulk Si, as discussed above, leading to eV for this scenario. Additionally, two H atoms could leave the passivating H layer and adsorb on the SiPc molecule [Fig. 3(f)]. The defect energy associated with a missing H atom can be approximated by the H-Si binding energy (PBE: eV, eV). A third possibility is the formation of a Si self-interstitial impurity, which can either form a complex with the substitutional Fe impurity or be isolated from it. One can see from Table 4 that all scenarios based on a removal of Fe from FePc with subsequent implantation into the H/Si matrix are energetically highly unfavorable, even when compared to typical room-temperature energies ( meV) and irrespective of the final Fe site, i.e., interstitial or substitutional. The situation remains unchanged if one tentatively lowers the PBE binding energy of FePc to a hybrid functional result (Table 3). In addition to the total energies of different initial and final states, kinetic barriers caused by transition states might inhibit the Fe exchange process.
The origin of the highly positive values of lies in the stability of the initial state, but also in the unfavorable final states considered so far. Let us now assume that two H atoms adsorb on a gas-phase FePc molecule [Figs. 3(d) and 4(a)]. If the Fe atom occupies a substitutional site after the reaction [Fig. 4(c)], the removed Si atom can be incorporated in the Pc ring, which results in SiPc with two additional H atoms above and below the molecular plane (SiPc-2H), which is very stable (Table 2). This scenario optimizes both and and leads to . If the Fe atom is integrated on an interstitial site [Fig. 4(d)], not SiPc-2H, but simply Pc remains after the reaction, resulting in as well.
Hence, the latter two mechanisms are statistically relevant (neglecting kinetic barriers). The crucial point is the adsorption of H atoms on the FePc molecule prior to the reaction at the H/Si surface. This can occur if, in addition to the FePc molecules, H atoms are evaporated from the crucible in the experiment, or if excess H atoms exist in the vicinity of the passivated surface. Moreover, a destabilization of FePc due to interactions with the H atoms in the passivating layer, as discussed above, facilitates the process.
We conclude from this analysis that subsurface doping of H/Si by room-temperature deposition of FePc molecules is possible, but the probability of the necessary steps in the reaction is low due to strongly bound Fe ion in FePc. More detailed investigations should be done in this field.
VI Summary
We studied the adsorption of iron phthalocyanine (FePc) on the passivated H/Si surface from first principles. According to our findings, the organic molecule is predominantly physisorbed with a distance to the surface of and an adsorption energy of eV, but also shows significant resonance with the underlying substrate. We used this paradigmatic case of a system in a mixed covalent-van-der-Waals-bonding state to compare the impact of different approaches to van-der-Waals interactions. (Spin-polarized) scanning tunneling microscopy (SP STM) images were simulated, selectively accessing different molecular orbitals via the applied bias voltage, similar to scanning tunneling spectroscopy. Comparison with experimental STM images revealed very good agreement. We found a considerable magnetic contrast exceeding in the SP STM images for and V.
In order to provide first insight into the mechanism behind a suggested subsurface doping strategy that involves room-temperature deposition of FePc on H/Si, we presented binding energies of different (transition metal) atoms in the center of the Pc ring, which particularly revealed that Fe is strongly bound (about eV). This surprising result was confirmed by different methodologies, including hybrid functionals for exchange and correlation, and holds for other transition metal centers as well. Despite this finding, we identified two feasible (exothermic) reactions by analyzing several distinct mechanisms to achieve subsurface Fe doping. Concomitantly, we highlighted the crucial role of a preceding destabilization of FePc, for instance, by pre-adsorbed H atoms, which subsequently strongly stabilize the final state of the reaction.
VII Acknowledgments
Computing time was granted by the Center for Computational Sciences and Simulation of the University of Duisburg-Essen.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jansen (2012) R. Jansen, Nat. Mater. 11 , 400 (2012) . · doi ↗
- 2Zhang et al. (2008) Z. Z. Zhang, B. Partoens, K. Chang, and F. M. Peeters, Phys. Rev. B 77 , 155201 (2008) . · doi ↗
- 3Küwen et al. (2009) F. Küwen, R. Leitsmann, and F. Bechstedt, Phys. Rev. B 80 , 045203 (2009) . · doi ↗
- 4Shaughnessy et al. (2010) M. Shaughnessy, C. Y. Fong, R. Snow, L. H. Yang, X. S. Chen, and Z. M. Jiang, Phys. Rev. B 82 , 035202 (2010) . · doi ↗
- 5Qian et al. (2006) M. C. Qian, C. Y. Fong, K. Liu, W. E. Pickett, J. E. Pask, and L. H. Yang, Phys. Rev. Lett. 96 , 027211 (2006) . · doi ↗
- 6Wu et al. (2007) H. Wu, P. Kratzer, and M. Scheffler, Phys. Rev. Lett. 98 , 117202 (2007) . · doi ↗
- 7Otrokov et al. (2011) M. M. Otrokov, A. Ernst, V. V. Tugushev, S. Ostanin, P. Buczek, L. M. Sandratskii, G. Fischer, W. Hergert, I. Mertig, V. M. Kuznetsov, and E. V. Chulkov, Phys. Rev. B 84 , 144431 (2011) . · doi ↗
- 8Ohno et al. (1996) H. Ohno, A. Shen, F. Matsukura, A. Oiwa, A. Endo, S. Katsumoto, and Y. Iye, Appl. Phys. Lett. 69 , 363 (1996) . · doi ↗