Excitation Variance Matching with Limited Configuration Interaction Expansions in Variational Monte Carlo
Paul J. Robinson, Sergio D. Pineda Flores, Eric Neuscamman

TL;DR
This paper introduces a variance matching method within variational Monte Carlo to accurately predict excitation energies in challenging molecules, especially when wave function flexibility is limited.
Contribution
It develops and tests a variance matching approach combining variational Monte Carlo and selective configuration interaction for better excitation energy predictions.
Findings
Effective at predicting excitation energies with limited wave function flexibility
Successful application to small difficult molecules like C3
Enhanced accuracy when combined with orbital optimization
Abstract
In the regime where traditional approaches to electronic structure cannot afford to achieve accurate energy differences via exhaustive wave function flexibility, rigorous approaches to balancing different states' accuracies become desirable. As a direct measure of a wave function's accuracy, the energy variance offers one route to achieving such a balance. Here, we develop and test a variance matching approach for predicting excitation energies within the context of variational Monte Carlo and selective configuration interaction. In a series of tests on small but difficult molecules, we demonstrate that the approach it is effective at delivering accurate excitation energies when the wave function is far from the exhaustive flexibility limit. Results in C, where we combine this approach with variational Monte Carlo orbital optimization, are especially encouraging.
Click any figure to enlarge with its caption.
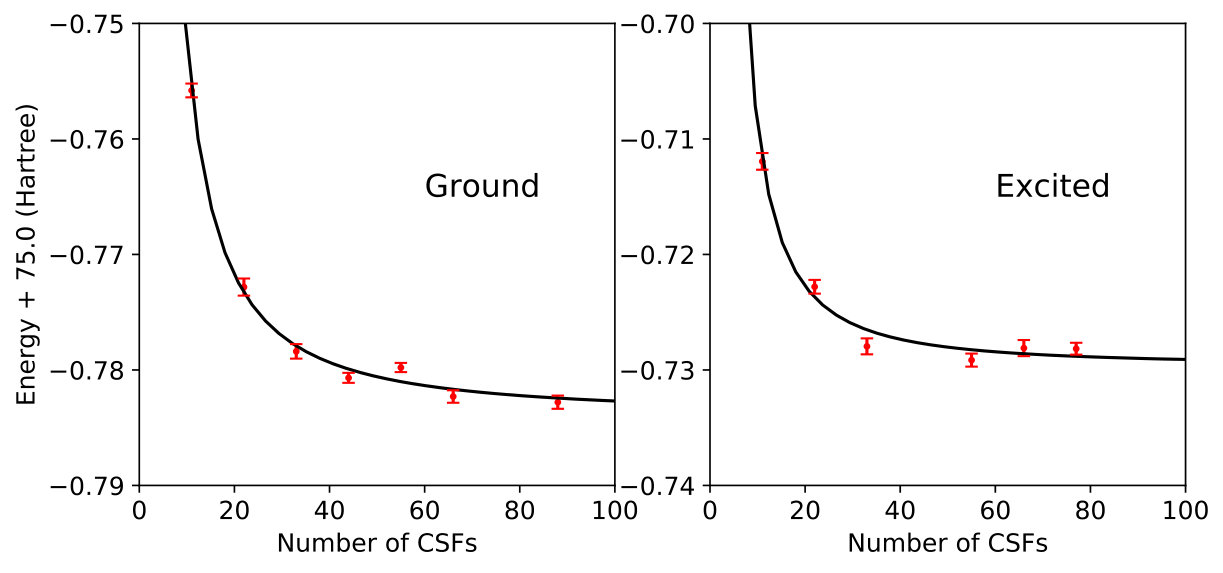 Figure 1
Figure 1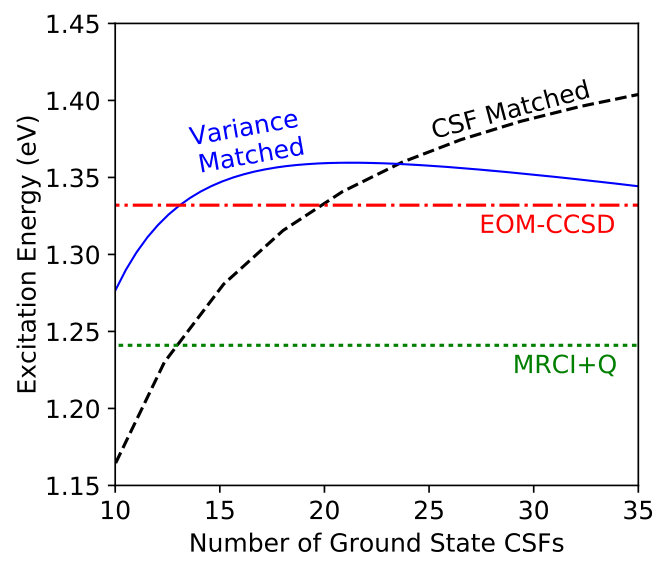 Figure 2
Figure 2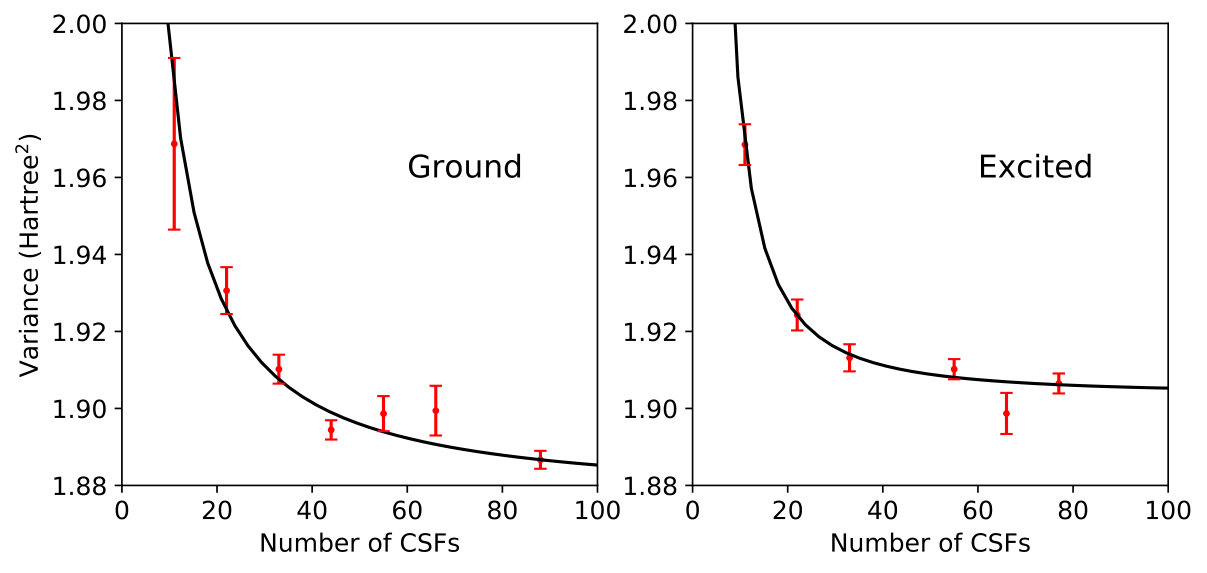 Figure 3
Figure 3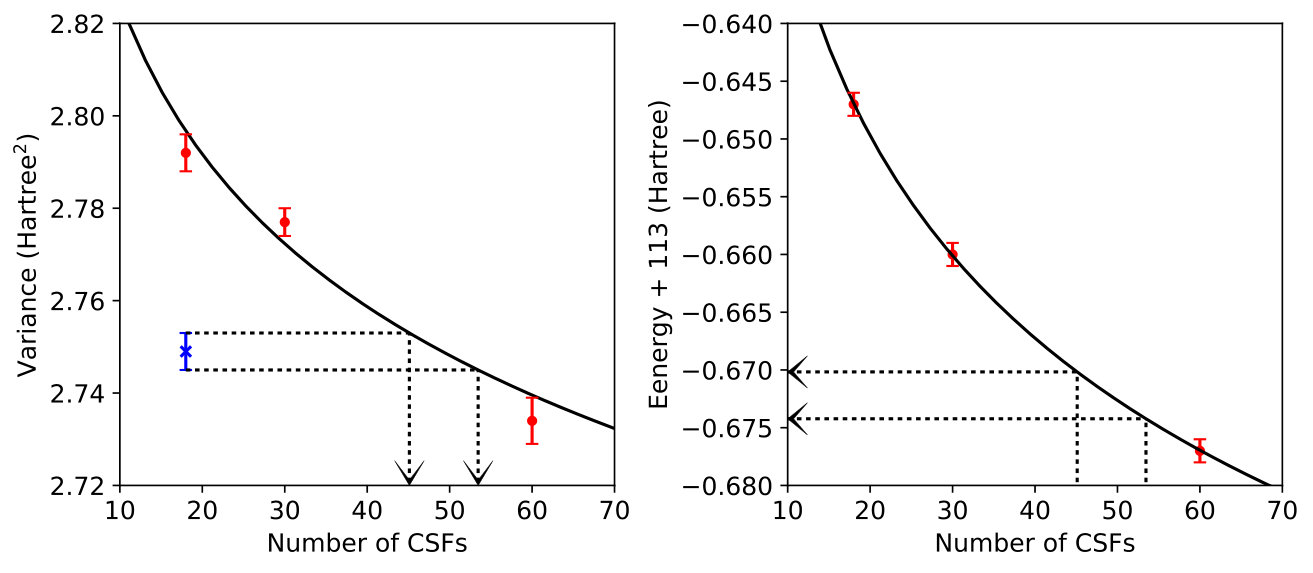 Figure 4
Figure 4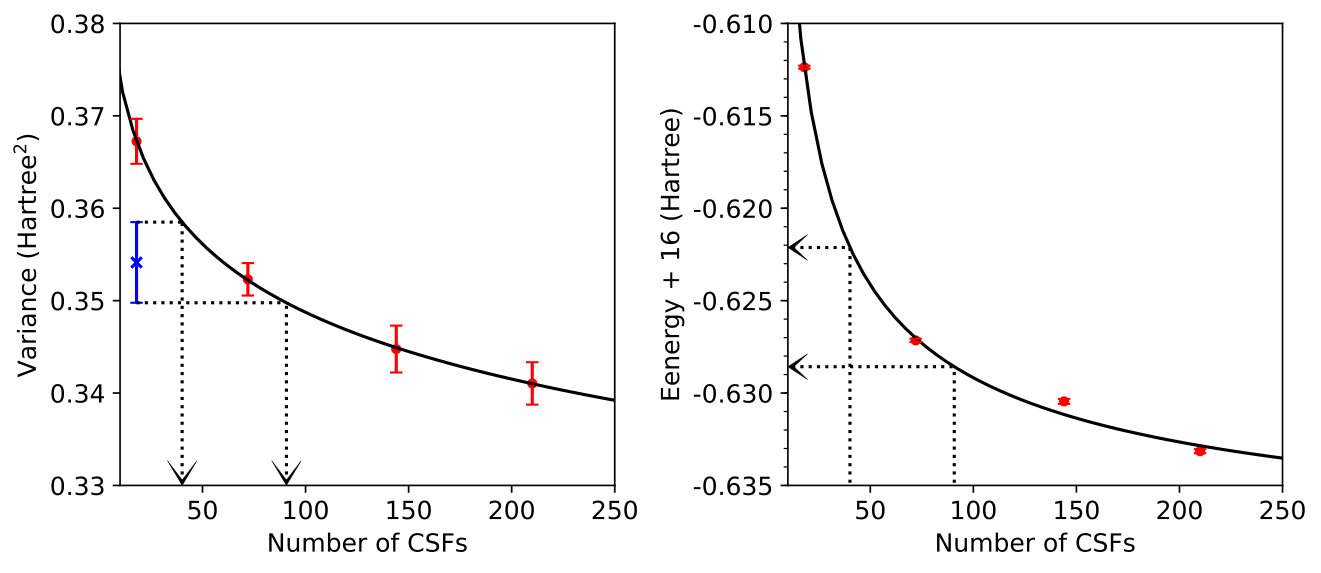 Figure 5
Figure 5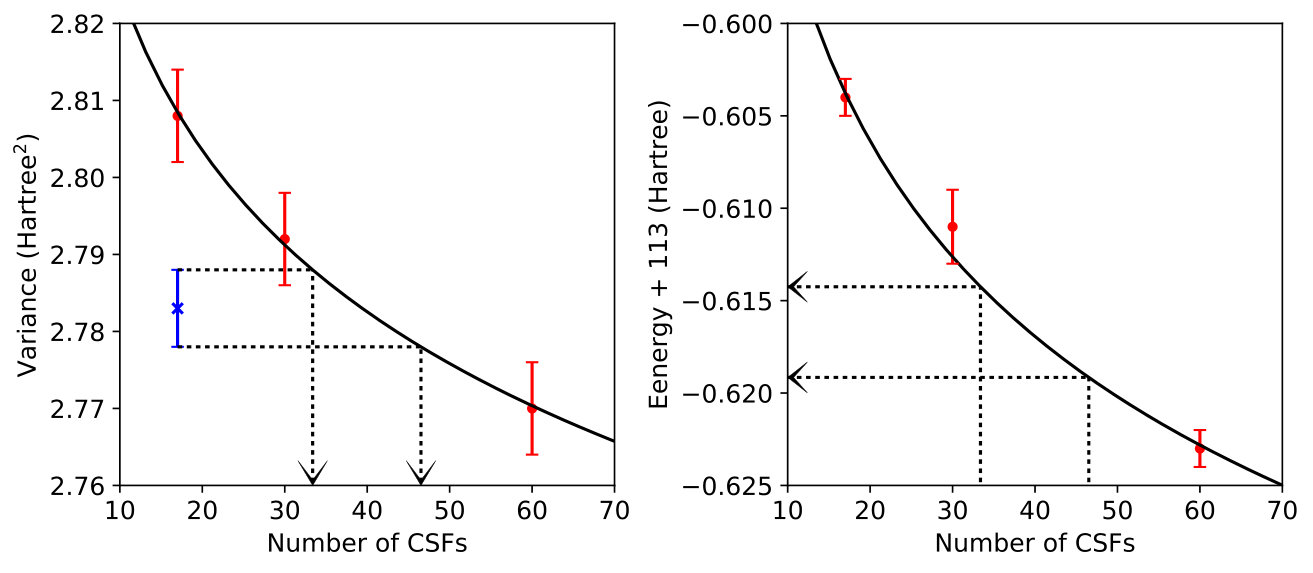 Figure 6
Figure 6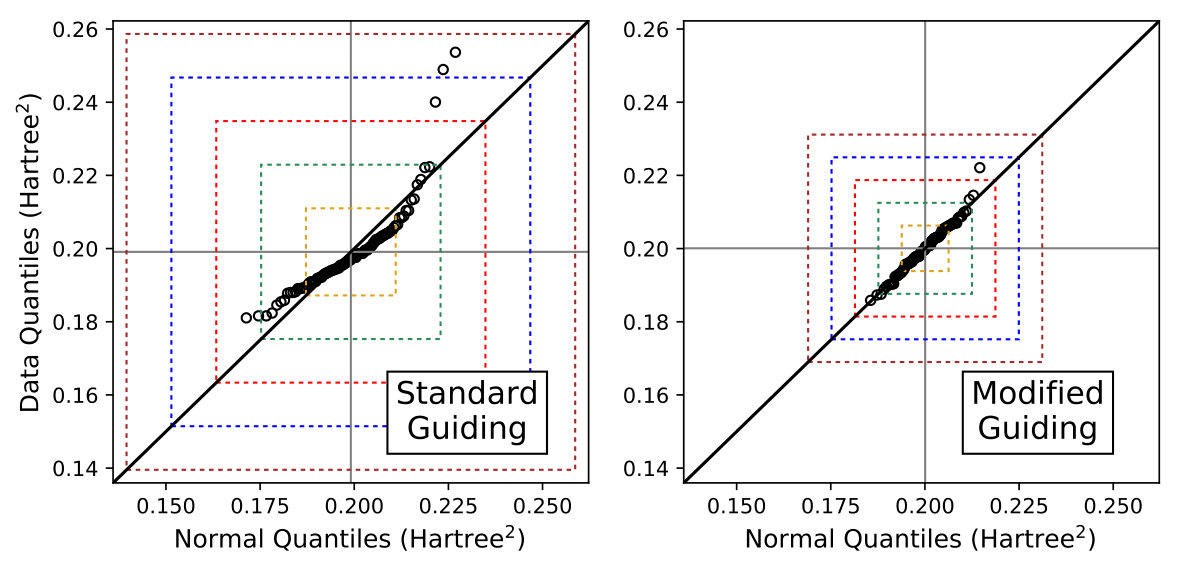 Figure 7
Figure 7| Method | Orbitals | h (eV) |
|---|---|---|
| EOM-CCSD | RHF | |
| CASSCF | CASSCF | |
| MRCI+Q | CASSCF | |
| -matched () | CASSCF | |
| CSF-matched () | CASSCF | |
| CSF-matched () | CASSCF |
| Method | Orbitals | h (eV) |
|---|---|---|
| EOM-CCSD | RHF | 3.402 |
| CASSCF | CASSCF | 3.303 |
| CASPT2 | CASSCF | 2.871 |
| MRCI+Q | CASSCF | 3.168 |
| CSF-matched | RHF | 4.46 0.05 |
| CSF-matched | CASSCF | 3.90 0.04 |
| -matched | RHF | 4.12 0.07 |
| -matched | CASSCF | 3.21 0.06 |
| -matched | VMC | 3.14 0.09 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Excitation Variance Matching with Limited Configuration Interaction Expansions in Variational Monte Carlo
Paul J. Robinson,1 Sergio D. Pineda Flores,2 and Eric Neuscamman*2,3,*111Electronic mail: [email protected]
1Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, USA
2Department of Chemistry, University of California, Berkeley, California 94720, USA
3Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
Abstract
In the regime where traditional approaches to electronic structure cannot afford to achieve accurate energy differences via exhaustive wave function flexibility, rigorous approaches to balancing different states’ accuracies become desirable. As a direct measure of a wave function’s accuracy, the energy variance offers one route to achieving such a balance. Here, we develop and test a variance matching approach for predicting excitation energies within the context of variational Monte Carlo and selective configuration interaction. In a series of tests on small but difficult molecules, we demonstrate that the approach it is effective at delivering accurate excitation energies when the wave function is far from the exhaustive flexibility limit. Results in C3, where we combine this approach with variational Monte Carlo orbital optimization, are especially encouraging.
I Introduction
Configuration interaction (CI) MolElecStruc , in which a many-electron wave function is approximated as a finite linear combination of Slater determinants, is among the most venerable and widely used approaches for predicting the effects of electron correlation on molecules’ properties and behavior. The use and development of CI-based theories continues today Sherrill:2005:importConfigs ; Ruedenberg:2009:deadwood ; booth2011CarbonDimer ; werner2011icmrci ; blunt2015krylov ; blunt2015semi ; Knowles:2015:compressive ; Alavi:2016:fciqmc_casscf ; Hoffmann:2016:ici ; Evangelista2016GuaranteedAcc ; Tubman:2016:asci ; Holmes:2016:heatBath ; sharma2017semistochastic ; zimmerman2017incremental ; ohtsuka2017selected despite their central shortcoming of having to choose between being size-inconsistent (in truncated forms) or having a cost that scales factorially with system size (in full (FCI) or complete active space (CAS) forms). Indeed, many researchers are willing to live with this flaw in light of the stability and robustness that arise from CI’s variational nature and straightforward systematic improvability.
Recently, these qualities have led to a resurgence in the study of selective CI (sCI), in which only a parsimonious selection of determinants is chosen, rather than the more traditional selection of all determinants below a given excitation level. sCI methods were originally developed bender1969studies ; huron1973iterative ; buenker1974individualized ; gouyet1976spin ; Evangelisti:1983hf ; harrison1991approximating at a time when high-accuracy treatments of weak correlation were being sought, a role that has now been more or less filled by coupled cluster theory. Barlett:2007:cc_rev More recently, there has been much renewed interest in sCI Sherrill:2005:importConfigs ; Ruedenberg:2009:deadwood ; Knowles:2015:compressive ; Hoffmann:2016:ici ; Evangelista2016GuaranteedAcc ; Tubman:2016:asci ; Holmes:2016:heatBath ; sharma2017semistochastic ; zimmerman2017incremental ; ohtsuka2017selected , especially in the context of treating strong correlation. While these approaches have made impressive progress, they do not remove the fundamental challenge that any CI, selective or otherwise, will lose its ability to systematically converge as system size increases and the number of determinants needed for a given level of accuracy quickly overwhelms available computing resources. Applications of CI, and indeed many other wave functions, to the wide class of chemical systems in which dozens or hundreds of atoms are involved thus require methods that can succeed without resorting to an exhaustive expansion of wave function flexibility.
In the context of predicting energy differences, one might instead rely on a cancellation of errors. In practice, however, this approach can be frustrated by the difficulty of achieving a balance between different states’ accuracies. At present, the most effective methods for achieving the necessary balance are those based on the state-averaged complete active space self-consistent field (CASSCF) approach, Werner:1985:mcscf ; Finley:1998:multistate_caspt2 but these rely on the exhaustive inclusion of all active space configurations and often the singles and doubles excitations out of the active space as well. Even difference dedicated CI, garcia1995iterative ; garcia1998treatment ; neese2003spectroscopy ; Zimmerman:2017:ddci_sf which aims to achieve balance without resorting to a full CAS, requires a relatively exhaustive inclusion of all wave function components related to the change between two states. In cases where even this more limited exhaustion of flexibility is not feasible, or where the physical insight required to choose the minimal CAS is lacking, a different route to achieving balance is required.
Here, we propose to achieve balanced treatments and accurate excitation energies in very short sCI expansions by employing the energy variance in combination with recent advances in state-specific optimization within variational Monte Carlo (VMC). Zhao:2016:dir_tar Like Evangelista’s “guaranteed accuracy” measure, Evangelista2016GuaranteedAcc is a direct measure of wave function accuracy. By varying different states’ sCI expansion lengths so that they are of equal accuracy as measured by , we will show that effective error cancellation and accuracies in the range of 0.1 or 0.2 eV can be achieved even for very short sCI expansions. Of course, this approach does not remove the ability to seek systematic convergence, as the variance-matched energy differences are guaranteed to converge to the exact result as the sCI expansions are enlarged. Instead, the approach allows for highly accurate results to be achieved even when one is far from the exact limit.
Certainly state-specific optimizations in VMC are not the only place where may be useful for achieving balance, and one can ask whether the approach is compatible with other Monte Carlo methods, such as full configuration interaction QMC booth2009fermion ; blunt2015semi and Monte Carlo CI. greer1995estimating ; greer1998monte In the former, an unbiased evaluation of may be possible based on the same two-population system used for density matrices. overy2014unbiased In the latter, it may be possible to estimate using a similar approach as is currently used to make perturbative corrections to sCI energies.
For the VMC approach pursued here, it is important to stress that two important advantages mitigate the fact that achievable expansion lengths are much shorter in VMC as compared to many other sCI approaches. First, Jastrow factors are known to significantly reduce the number of Slater determinants required for an accurate CI expansion. UmrTouFilSorHen-PRL-07 Second, these multi-Slater Jastrow (MSJ) wave functions (whose computational efficiency has recently been greatly enhanced miguel:2011:table ; Scuseria:2012:msj ; Filippi:2016:msj_gradients ; assaraf2017optimizing ) can be used to provide nodal surfaces for diffusion Monte Carlo (DMC), Foulkes:2001:qmc_review which is highly effective at recovering the effects of weak correlation that can be missed by limited sCI expansions. Crucially, both MSJ wave functions assaraf2017optimizing and DMC benali2014application have been demonstrated to scale to systems with over 100 atoms, implying that the advantages of variance matching should scale to a wide variety of chemical applications. In this paper, we will take a first step towards such applications by exploring the practical considerations that arise in working with this idea and by establishing its accuracy in a handful of small systems where high level benchmarks are available.
II Theory
II.1 Estimating the variance
The energy variance,
[TABLE]
can be estimated by Monte Carlo integration following the placement of a resolution of the identity in between the two factors of ,
[TABLE]
Defining the local energy as and letting , this integral can be written as the square deviation of the local versus total energy averaged over the wave function’s probability distribution,
[TABLE]
Estimating by VMC thus amounts to sampling from the wave function’s probability distribution and averaging this square energy difference, which requires that exactly the same information be extracted from the wave function ansatz as in a standard VMC estimation of the energy . Foulkes:2001:qmc_review ; Umrigar:2005:lm Crucially, the resolution of the identity between the two powers of ensures that can be estimated without ever squaring the Hamiltonian operator explicitly. This advantage is the reason that VMC can access the variance more easily than more traditional methods in quantum chemistry, such as configuration interaction MolElecStruc or coupled cluster theory. Barlett:2007:cc_rev Indeed, given that VMC energy estimates also require evaluating at a large number of sampled configurations, an estimate of the variance is typically available “for free” during the normal use of VMC for energy evaluation and minimization. However, as we discuss in Section II.4, it can be statistically advantageous to estimate the integral in Eq. (2) via Monte Carlo integration over a different probability distribution than that of the wave function.
II.2 Targeting excited states
To optimize a MSJ expansion for a particular Hamiltonian eigenstate, we minimize the recently introduced Zhao:2016:dir_tar target function
[TABLE]
(in which the second equality follows from ) with respect to both the nonlinear Jastrow factor variables and the CI expansion’s linear coefficients. Similar to the energy function, whose global minimum is the ground state, this function has its global minimum at the energy eigenstate whose energy is immediately above the target energy , allowing it to target either ground states or excited states. As with the variance , the denominator of may be evaluated efficiently Zhao:2016:dir_tar by resolving an identity between the two powers of and employing Monte Carlo integration. As we have done in previous uses of this excited state target function, Zhao:2016:dir_tar ; Neuscamman:2016:var ; Zhao:2017:blocked_lm we ensure a unique choice for by adjusting it to minimize for the state in question.
II.3 Balancing the states
At present, the most effective approaches for achieving a balanced description of ground and excited states are exhaustive in nature. For example, CAS-based approaches such as MRCI+Q werner2011icmrci rely on the balance provided by constructing the active space to contain all configurations that make major contributions to either state. While these approaches are highly effective, this type of exhaustive strategy is not feasible in larger systems, for which it is natural to ask the question: what is the best alternative? One could try to give “equal” flexibility to both states’ ansatzes in a sCI by allowing them the same number of configuration state functions (CSFs), although there is no reason to assume that this CSF-matched approach will yield states with similar energy errors. Instead, we would prefer to seek cancellation of error by matching a quantity that is actually related to the error in a wave function.
In this study, we will therefore explore the possibility of improving accuracy in energy differences by insisting that such differences be taken between wave functions with equal variance, as is a direct measure of wave function accuracy. We take as our wave function a MSJ expansion,
[TABLE]
in which each is a different CSF — i.e. a spin adapted linear combination of determinants with the same spinless occupation pattern — and the factor outside the sum is the Jastrow factor. Foulkes:2001:qmc_review In this study, the Jastrow factor is taken as
[TABLE]
in which is the distance between the th electron and th nucleus, the distance between the th and th electrons, and and are one-dimensional functions represented by optimizable 10-point splines. Note that we use two different functions for for same-spin versus opposite-spin electron pairs and enforce the relevant electron-electron cusp condition in both cases.
Variational optimization of Eq. (5) via the minimization of Eq. (4) will produce one value of the variance for each length chosen for the CSF expansion. To assist in comparing the variances (or energies) of different states, we will often interpolate by fitting this (or ) versus distribution to a power law decay of the form
[TABLE]
in which , , and are chosen by a least-squares fit to the values of the optimized wave functions for a handful of different CSF expansion lengths . This approach allows us to avoid tedious searches for the precise expansion length at which the excited state variance most closely matches that of the ground state.
Using these interpolating functions, our approach to variance matching is the following. First, we optimize the ground state for a small CSF expansion and evaluate its variance. We then optimize the excited state in question for a range of expansion lengths and interpolate via Eq. (7) to find the excited state expansion length for which the ground and excited states’ variances match. We then employ an analogous interpolation of the excited state energy to produce the variance-matched excitation energy.
II.4 Modified guiding function
Unfortunately, the statistical uncertainty of VMC estimates of the variance are not well defined Trail:2008:heavy_tail ; Trail:2008:alt_sampling when using the standard importance sampling function for the Monte Carlo integration evaluation of Eq. (2). To understand why this comes about, consider the nodal surface of the wave function. Near a node, the wave function is by definition heading to a zero value, but its second derivative, which contributes to the local energy via the kinetic energy term, need not be zero. It is thus possible for
[TABLE]
as approaches the nodal surface. While the average energy and its variance remain finite despite this divergence, the variance of the variance does not, and so the central limit theorem cannot be used to estimate the statistical uncertainty in the estimated value of the variance. Here, we will address this issue by modifying the guiding function so as to avoid problematic divergences (see Appendix A for an explicit example of how such modifications can help).
Specifically, we replace with the guiding function
[TABLE]
in order to avoid the divergences in the local energy that cause an infinite variance of the variance. To keep this distribution as close to as possible when is not near a node (which is advantageous as it allows us to approach closer to the zero variance principle) the denominator in Eq. (9) works to smoothly “switch off” the adjustment as the logarithm of the wave function rises above the threshold . While the magnitude of will of course depend on the parameters , , and , it will be finite so long as and . We note that, in practice, evaluating as part of the sampling function is expensive, and so in future it will be desirable to test other less expensive alternatives for modified sampling functions. For the tests in the present study, however, we find this sampling function to be affordable.
III Results
III.1 Computational Details
VMC calculations were performed in a development version of QMCPACK qmcpack_1 ; qmcpack_2 , with the molecular orbital basis and the choice of CSFs derived from restricted Hartree-Fock (RHF), complete active space self-consistent field (CASSCF) and CI calculations in GAMESS gamess1 as described for each individual molecule in Appendix B. Equation of motion coupled cluster with singles and doubles (EOM-CCSD), multi-reference CI with Davidson correction (MRCI+Q), and complete active space second order perturbation theory (CASPT2) calculations were carried out with Molpro. MOLPRO-WIREs
In all cases, the CSFs used for a given MSJ expansion were taken as the first CSFs from a CISDTQ calculation. While this is of course not the most efficient way to generate a short sCI expansion, it is sufficient for our purposes of testing the efficacy of variance matching. The CSF coefficients and Jastrow variables of each MSJ wave function were optimized by minimizing , with at first held fixed to ensure convergence to the desired state but then adjusted so as to minimize for that state (see Reference Zhao:2016:dir_tar for details). The sample length and number of optimization steps were chosen so as to ensure the statistical uncertainty in the optimized energy was converged to within one or two milliHartrees. Standard deviations of statistically estimated quantities are displayed in the data below. In the case of Figure 1 where the central limit theorem does not apply, it should be understood that although the error bars have been evaluated using the typical formula, the underlying statistics cannot be assumed to be Gaussian.
In all cases, the electron-nuclear and electron-electron Jastrow factors were parameterized as exponentials of 10-point splines with 5-Bohr and 10-Bohr cutoffs, respectively. Nuclear cusps were enforced by augmenting the orbitals Needs:2005:cusps_in_orbs , while electron-electron cusp conditions Foulkes:2001:qmc_review were enforced via the electron-electron Jastrow. For system-specific computational details, such as geometries and basis sets, we refer the reader to Appendix B.
III.2 C2
We begin our investigation of variance matching in the carbon dimer by optimizing a series of MSJ wave functions with increasingly large CSF expansions for both the ground and first excited singlet state. We intentionally restrict ourselves to relatively short expansions with no more than 100 CSFs so as to simulate a situation in which results cannot be converged by the brute-force expansion of the wave function, even though this would of course be possible in a system as small as C2. As shown in Figures 1 and 2, increasing the CSF length has the expected effect of reducing the variance and energy for both states.
Note that we completed our studies of C2 and allene before implementing the adjusted importance sampling function of Eq. (9), and so these systems’ results are based on variance estimates using the standard importance sampling. As expected, this appears to have had a deleterious effect on the uncertainty in the variance, but in both cases it was nonetheless possible to perform reasonable fits for the purposes of variance matching. We will see that the variance statistics are much better behaved in H4 and C3, where we have utilized our modified importance sampling.
Despite the somewhat noisy data, we can use the fits to Eq. (7) to produce an estimate of what the excitation energy would be when the excited state MSJ expansion length is chosen so that it has the same variance as the ground state. We plot these results in Figure 3 alongside the excitation energies that result when both states are restricted to have the same number of CSFs, which, despite enforcing equality in flexibility, is ineffective at balancing accuracy as the expansion length needed to handle the effects of correlation in different states is of course not uniform. Indeed, we see that the size of the ground state CSF expansion has a smaller effect on the predicted excitation energy under variance matching, with predictions all falling within 0.09eV of each other for ground state CSF lengths between 10 and 35 CSFs. If instead we match the number of CSFs in each state, the predicted excitation energy changes by 0.24eV over this range. Furthermore, the variance-matched results appear to converge more rapidly towards agreement with the benchmark EOM-CCSD and MRCI+Q results as the number of ground state CSFs is increased. Thus, as hoped, the use of variance matching for this excitation appears to help offset the fact that the two states require significantly different numbers of CSFs to reach descriptions of equivalent accuracy.
III.3 Allene
The second system we investigate is allene, C3H4. Unlike C2, in which the ground state itself is quite multi-reference, allene presents us with a case in which the ground and first excitation have significantly different degrees of multi-reference character. The ground state of allene is dominated by the closed shell determinant: its CI coefficient is 0.96 in CASSCF and only 3 CASSCF CI coefficients are greater than 0.001. In contrast, the first excited state has 14 CASSCF CI coefficients above this threshold, making allene an excellent candidate for a system in which variance matching should be helpful.
As with C2, we limited ourselves to very short CSF expansions and found that when the ground state was optimized with a 22-CSF MSJ, the variance was 3.44 Hartree2. After some initial trial-and-error and interpolation, we found that the same variance results for the excited state when a 66-CSF MSJ expansion is employed. In Table 1, we see that when MSJ expansions with the same number of CSFs are used for both the ground and excited state, the excitation energy is overestimated as compared to the EOM-CCSD and MRCI+Q results, which is what we would expect given the more multi-reference character of the excited state. CSF-matched results do improve as the number of CSFs is increased from 22 to 66, but we see that it is more accurate to evaluate the excitation energy based on a ground state with 22 CSFs and an excited state with 66, which as stated above produces states with equal values of Hartree2. Thus, as expected, variance matching for this excitation reduces the ground state bias that originates from having a more multi-reference excited state.
III.4 H4
At this point in our investigation, we decided that although variance matching looked promising, the poor statistics of the variance should be addressed before proceeding further. To this end, we implemented importance sampled Monte Carlo integration based on Eq. (9) in our development version of QMCPACK. To validate that the infinite variance of the variance issue was satisfactorily resolved and the statistics of the variance were now normal as per the central limit theorem, we performed normality tests for variance data on a simple system of two nearby hydrogen molecules, one with a near equilibrium bond length and the other slightly stretched, a system we will refer to as H4. After optimizing the MSJ ansatz for the ground state, we interrogated the statistical distribution of its variance by estimating it with multiple, independent samples of electron configurations, the results of which are displayed in Figure 4.
The statistical distribution of is clearly skewed away from normal when using the standard guiding function, confirming that the central limit theorem is not in effect, as expected. Trail:2008:heavy_tail This conclusion is further buttressed by the observation that two out of the 100 estimates taken with lie more than four standard deviations away from the mean. On average, normally distributed data would only show two 4- events like this after 31,574 estimates, and here we see two after just 100.
Using the modified guiding function, the variance estimates match a normal distribution much more closely, and we observe no 4- events. We do see one 3- event among the 100 estimates, compared to an expectation that, on average, one 3- event will occur in every 370 estimates for normally distributed data. This deviation is much more believably either a statistical fluctuation or the effect of the fact that our Monte Carlo samples of configurations are finite and so we may not yet have converged to the perfectly Gaussian limit. In addition to becoming more normal in their distribution, the statistical uncertainties in the variance are also smaller with the modified guiding function, at least if we measure by their standard deviation. Thus, as previously predicted, Trail:2008:heavy_tail ; Trail:2008:alt_sampling it appears that we can indeed resolve the infinite variance of the variance dilemma via a modification to the importance sampling function that we employ in our Monte Carlo integration.
III.5 C3
Our final test system in this preliminary investigation of variance matching is C3, the linear carbon trimer, in which we use the improved variance statistics granted by the modified guiding function to help resolve the importance of the choice of molecular orbital basis. In a setting where a sCI expansion can be converged with respect to CSF number, the molecular orbital basis will of course not matter. However, in the more practical case we are considering here, in which converging the expansion is prohibitively expensive, this guarantee cannot be relied upon. We address this issue by testing variance-matching in three sets of molecular orbitals: RHF-optimized orbitals, CASSCF-optimized orbitals, and VMC-optimized orbitals. Note that for the latter case, we have optimized the orbitals separately for the ground state and the first excited state (see Appendix B) using a recently-implemented combination of our direct targeting method and the efficient MSJ orbital optimization approach of Filippi and coworkers. Filippi:2016:msj_gradients ; assaraf2017optimizing
For each choice of basis, we first performed a ground state MSJ optimization with a short CSF expansion. Next, we performed excited state optimizations with different CSF expansion lengths so that we could estimate by interpolation the excited state expansion length (and its corresponding energy) at which the ground and excited state variances would match. This process and its results are displayed graphically in Figures 5-7 for our three different choices for the molecular orbitals. Before analyzing the effects of variance matching, it is worth noting that, thanks to the improved guiding function, the variance uncertainties in this case are smaller than they were in C2, despite the 50% larger system size.
As anticipated, Table 2 reveals that the choice of the molecular orbitals matters when taking a variance-matching approach to excitation energy prediction. We see that results using RHF orbitals are quite poor, which is to be expected due to the bias they create in favor of the ground state. On the other hand, excitation energies based on CASSCF or VMC-optimized orbitals are within statistical uncertainty of the benchmark (cc-pVTZ, full-valence-CAS) MRCI+Q results. Although one cannot expect to have access to CASSCF orbitals in large systems, it is possible assaraf2017optimizing to produce VMC-optimized orbitals, making their excellent performance in conjunction with variance matching and short CSF expansions an extremely promising result. As a final note, and as we saw in other cases, CSF-number-matched results are less accurate, reinforcing the point that providing equal amounts of wave function flexibility is no guarantee that the errors in different states will be similar. Instead, it proves far more effective to ensure that the states are balanced in terms of a rigorous error metric, such as .
IV Conclusions
Extending the success of sCI methods into regimes where exhaustive convergence is not possible would greatly expand their utility in chemical investigations. Towards this end, we have developed and tested a strategy for excitation energies based on variational Monte Carlo’s ability to use the energy variance to balance the descriptions of different states. In tests on C2, allene, and C3, our variance-matched results are within 0.1, 0.2, and 0.1 eV, respectively, of cc-pVTZ MRCI+Q excitation energies. These accuracies are achieved despite using ground state selective configuration interaction expansions that contain fewer than 40 configuration state functions and are thus very far from the exhaustive flexibility limit. As multi-Slater Jastrow optimizations can now handle hundreds of thousands of determinants and molecules containing over 100 atoms, Filippi:2016:msj_gradients ; assaraf2017optimizing these preliminary results suggest that variance matching could play a major role in future high-accuracy work in molecular excited states.
To make further progress towards this goal, it will be important to focus improvement on the most important aspects of the methodology. As we showed in this study, statistical estimates of the variance are greatly improved by using a non-standard guiding function for the Monte Carlo integration that, although it has the same scaling with system size, is more expensive to work with than the standard guiding function. It would thus be beneficial to pursue more sophisticated guiding functions that are both inexpensive and effective at reducing uncertainty in variance estimates. Another avenue of investigation that will clearly be important is the choice of selective CI scheme. While many options exist, none are currently native to quantum Monte Carlo, by which we mean that they do not take account of the Jastrow correlation factor when making decisions about which configurations to use. Developing selection schemes that couple more closely to quantum Monte Carlo is therefore highly desirable. By pursuing improvements in these areas, and by pushing to larger systems and more aggressive determinant expansions, we look forward to future explorations of how variance matching can complement existing methods in achieving balanced and accurate treatments of molecular excited states.
V Acknowledgements
The authors acknowledge funding from the Office of Science, Office of Basic Energy Sciences, the US Department of Energy, Contract No. DE-AC02-05CH11231. Parallel development and debugging of the software for nodeless guiding functions was performed using the Berkeley Research Computing Savio cluster. Molecular calculations were run at the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Appendix A Modified Guiding Example
To see how the breakdown of the central limit theorem comes about and how this difficulty can be addressed by a modified guiding function, consider a two-dimensional integral
[TABLE]
[TABLE]
which we might try to evaluate via importance-sampled Monte Carlo integration. For a given choice of normalized importance-sampling function , we have
[TABLE]
The variance for this choice of sampling function is
[TABLE]
and the variance of the variance is
[TABLE]
Given that the function decays rapidly, an excellent approximation to the integral can be had by choosing to be uniform on the square , . This produces the mean, variance, and variance of the variance
[TABLE]
If instead we choose our sampling function as
[TABLE]
with the normalization constant, we have
[TABLE]
Although this importance sampling function reduces the variance, which will in turn reduce the statistical uncertainty in , it creates a singularity in at the origin. For , this singularity is integrable. For , which contains a higher power of , it is not. We are therefore unable to rely on the central limit theorem in this case to analyze the uncertainty in a Monte Carlo integration estimate of the variance, as the variance of the variance is not finite.
This issue is readily resolved via a small modification to the sampling function,
[TABLE]
which removes the singularity in while only slightly altering its values away from the origin. The result,
[TABLE]
is that the variance is essentially unchanged, but the variance of the variance is now finite as there are no longer any singularities. Thus, in this example, we see that a small change to the sampling function that removes singularities while otherwise leaving the function more or less unaltered leads to a qualitative improvement in statistical estimation. For a rigorous analysis of these issues in VMC, we refer the reader to the work of Trail. Trail:2008:heavy_tail ; Trail:2008:alt_sampling
Appendix B Computational Details
C2: The C2 bond length was chosen to be 1.2425146399 Å. The basis set was chosen as cc-pVTZ for both the CASSCF and the high-level reference calculations. The molecular orbitals were obtained from an (8e,8o) state-averaged CASSCF for the ten lowest lying singlet states. The configuration list from which truncations were taken for MSJ expansions was generated via a CISDTQ calculation in which the 1s orbitals were held frozen and excitations were allowed among the 3rd through 22nd CASSCF orbitals.
Allene: The geometry for allene (C3H4),
C 0. 000000000 -0. 000000000 -0. 000000012
C 0. 000000000 -0. 000000000 2. 481842730
C 0. 000000000 0. 000000000 -2. 481842716
H 1. 756493809 0. 000000000 -3. 532793008
H -1. 756493809 0. 000000000 -3. 532793008
H 0. 000000000 1. 756493826 3. 532792993
H 0. 000000000 -1. 756493826 3. 532792993
was chosen based on a CCSD(T) optimization with the cc-pVTZ basis set in Molpro. The CASSCF orbitals were obtained from a 4-state-averaged CASSCF(10e,16o) calculation in GAMESS with the cc-pVTZ basis. The CSFs used for the MSJ expansion were generated from a 4-state CISDTQ calculation with the 1s orbitals on carbon frozen and excitations allowed among the 4th through 20th CASSCF orbitals.
H4: This system consists of two nearby hydrogen molecules, one with its bond somewhat stretched so as to induce a degree of correlation in the ground state, for which we optimized a 2-CSF MSJ wave function. The basis set was STO-3G, with the molecular orbital basis for the CI expansion taken as the RHF canonical orbitals. The atomic coordinates are:
0. 0000000000 0. 0000000000 0. 0000000000
1. 8897259877 0. 0000000000 0. 0000000000
0. 0000000000 0. 0000000000 2. 8345889816
0. 0000000000 0. 0000000000 5. 6691779632
C3: The carbon atoms were arranged in a line with 1.302 Å between atoms. All QMC calculations used the cc-pCVTZ basis set Dunning:1989:basis_for_corr to help reduce variance in the core region, while reference calculations were performed in the more typical cc-pVTZ basis. The reference CASSCF, CASPT2, and MRCI+Q calculations used a (12e,12o) “full valence” active space. In EOM-CCSD, CASPT2, and MRCI+Q, the 1s orbitals were set as an inactive frozen core.
To generate our multi-Slater expansion, we employed molecular orbitals from either RHF or an equally-weighted CASSCF state average of the first three singlet states in an (8e,10o) active space. For a given choice of CSF number , the MSJ’s CI expansion was constructed using the CSFs with the largest coefficients after a single-reference CISDTQ calculation (in either the RHF or CASSCF orbitals) in which the 1s orbitals were held frozen and excitations were allowed among the 4th through 27th molecular orbitals. Note that for the ground state, we used 17 CSFs in the RHF orbital case and 18 CSFs in the CASSCF orbital case, as these choices avoided splitting degenerate CSFs between the MSJ’s used and unused CSF sets.
The VMC optimized molecular orbitals were found by simultaneously optimizing the orbitals, Jastrow variables, and CI coefficients for 18-CSF MSJ wave functions for the ground and excited states separately. To aid convergence in these optimizations, we have replaced the 1s electrons with an effective core potential carbon_pseudopotential in all calculations involving VMC-optimized orbitals. For consistency, we took our CSFs in this case from the closest possible CISDTQ, which was generated in the same way as for the CASSCF orbital case, except that the effective core potential was employed.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1(1) T. Helgaker, P. Jøgensen, and J. Olsen, Molecular Electronic Structure Theory , John Wiley and Sons, Ltd, West Sussex, England, 2000.
- 2(2) M. L. Abrams and C. D. Sherrill, Chem. Phys. Lett. 412 , 121 (2005).
- 3(3) L. Bytautas and K. Ruedenberg, Chem. Phys. 356 , 64 (2009).
- 4(4) G. H. Booth, D. Cleland, A. J. W. Thom, and A. Alavi, J. Chem. Phys. 135 , 084104 (2011).
- 5(5) K. R. Shamasundar, G. Knizia, and H.-J. Werner, J. Chem. Phys. 135 , 054101 (2011).
- 6(6) N. S. Blunt, A. Alavi, and G. H. Booth, Phys. Rev. Lett. 115 , 050603 (2015).
- 7(7) N. S. Blunt et al., J. Chem. Phys. 142 , 184107 (2015).
- 8(8) P. J. Knowles, Mol. Phys. 113 , 1655 (2015).