Monitoring Nonadiabatic Avoided Crossing Dynamics in Molecules by Ultrafast X-Ray Diffraction
Markus Kowalewski, Kochise Bennett, Shaul Mukamel

TL;DR
This paper demonstrates how ultrafast X-ray diffraction can monitor nonadiabatic avoided crossing dynamics in molecules, revealing electronic and nuclear structural changes with detailed signal contributions and simulations of sodium fluoride.
Contribution
It introduces a method to distinguish different scattering contributions in time-resolved X-ray diffraction during nonadiabatic dynamics, including electronic coherences and transition charge densities.
Findings
Elastic scattering reveals molecular geometry changes.
Electronic coherences contribute weaker but spatially informative signals.
Simulations of sodium fluoride's nonadiabatic process validate the approach.
Abstract
We examine time-resolved X-ray diffraction from molecules in the gas phase which undergo nonadiabatic avoided-crossing dynamics involving strongly coupled electrons and nuclei. Several contributions to the signal are identified, representing (in decreasing strength) elastic scattering, contributions of the electronic coherences created by nonadiabatic couplings in the avoided crossing regime, and inelastic scattering. The former probes the charge density and delivers direct information on the evolving molecular geometry. The latter two contributions are weaker and carry spatial information of the transition charge densities (off-diagonal elements of the charge-density operator). Simulations are presented for the nonadiabatic harpooning process in the excited states of sodium fluoride.
Click any figure to enlarge with its caption.
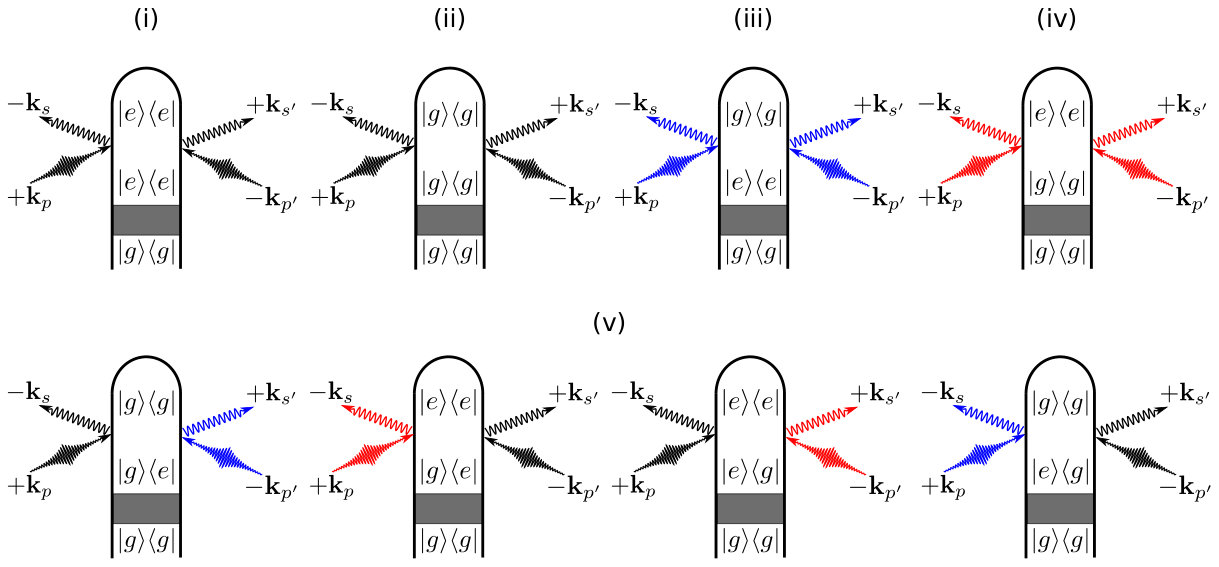 Figure 1
Figure 1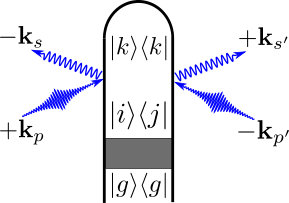 Figure 2
Figure 2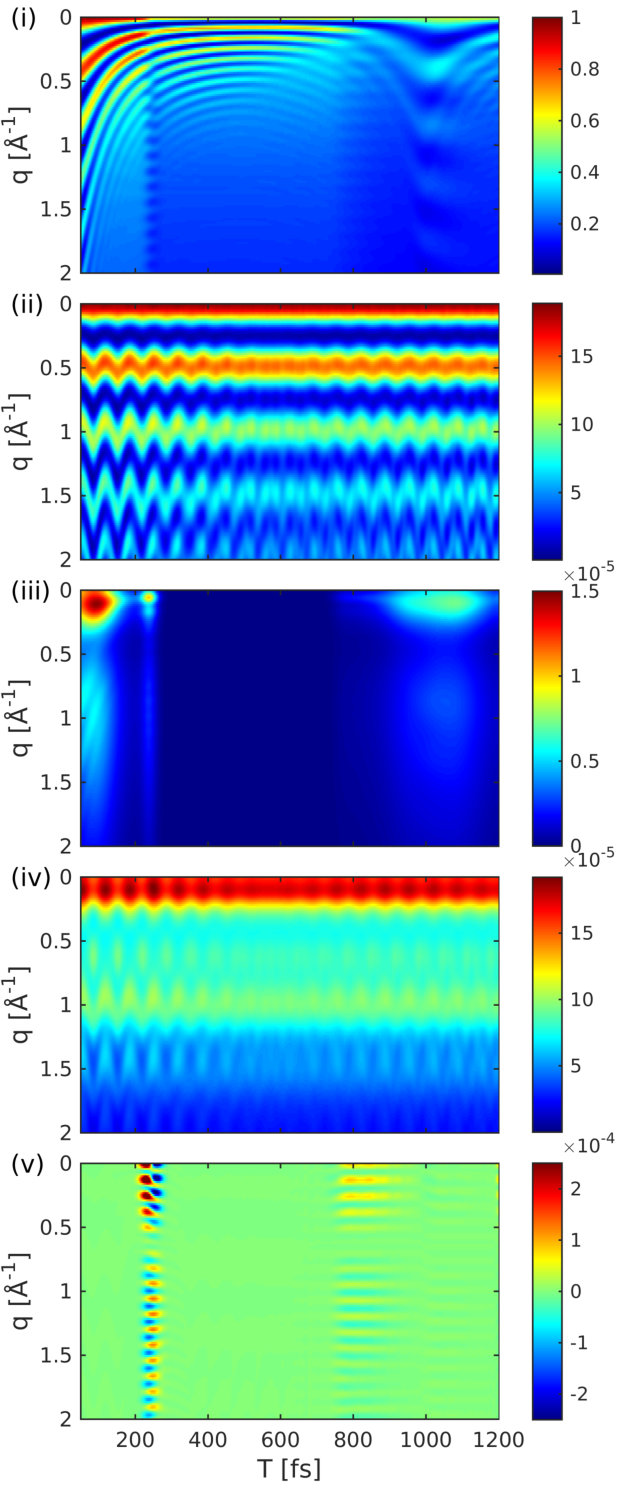 Figure 3
Figure 3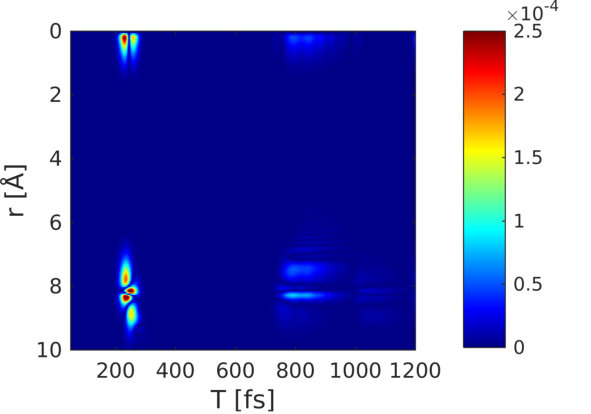 Figure 4
Figure 4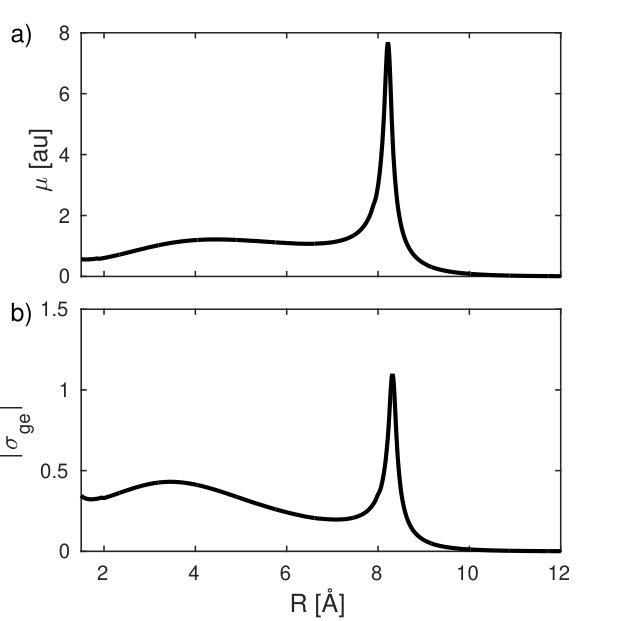 Figure 5
Figure 5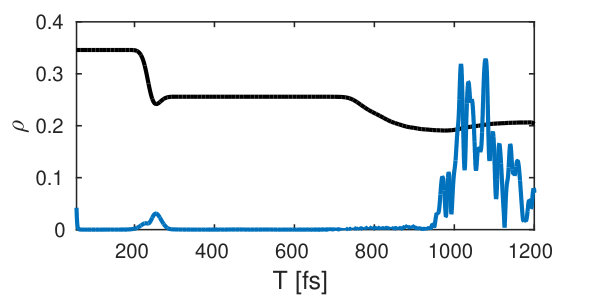 Figure 6
Figure 6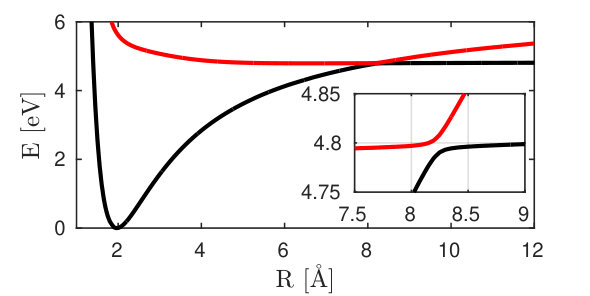 Figure 7
Figure 7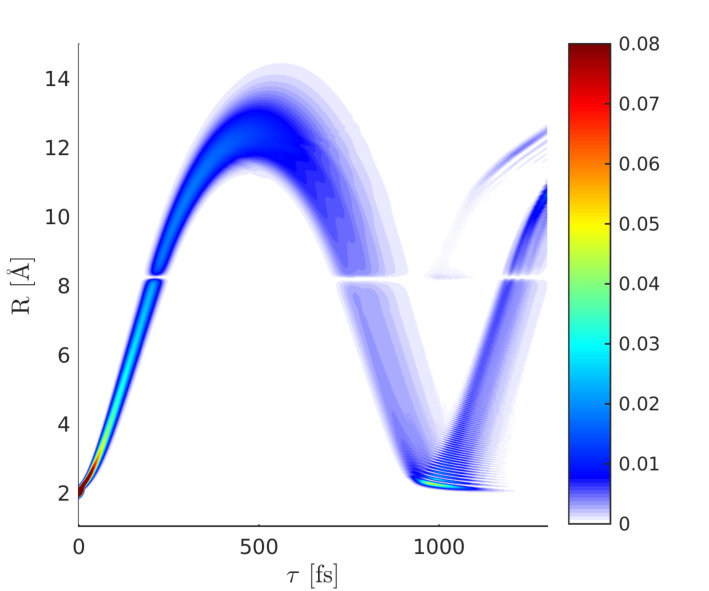 Figure 8
Figure 8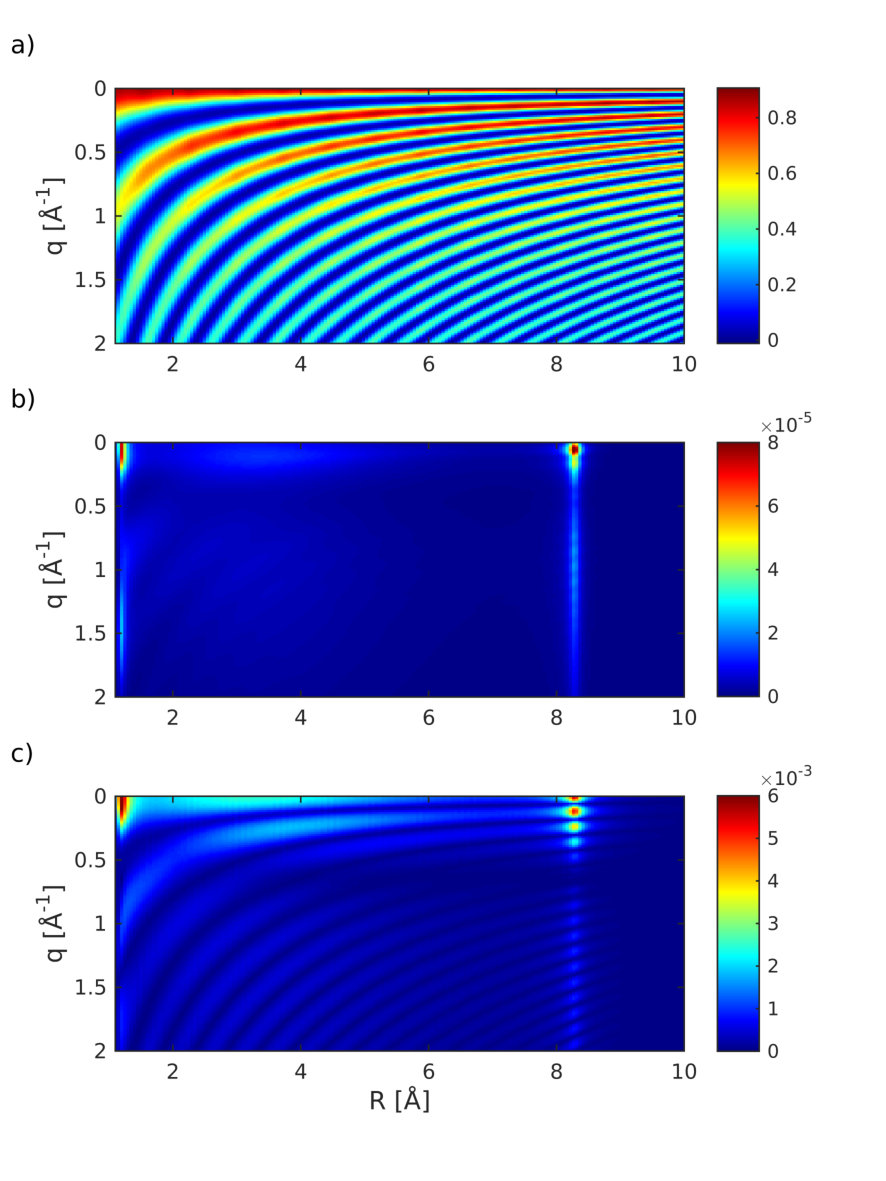 Figure 9
Figure 9Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
††thanks: These authors contributed equally to this manuscript
Monitoring Nonadiabatic Avoided Crossing Dynamics in Molecules by Ultrafast X-Ray Diffraction
Markus Kowalewskia,b,†
Kochise Bennettb
Shaul Mukamelc
Chemistry Department, University of California, Irvine, California 92697-2025, USA
Department of Physics and Astronomy, University of California, Irvine, California 92697-2025, USA
Abstract
We examine time-resolved X-ray diffraction from molecules in the gas phase which undergo nonadiabatic avoided-crossing dynamics involving strongly coupled electrons and nuclei. Several contributions to the signal are identified, representing (in decreasing strength) elastic scattering, contributions of the electronic coherences created by nonadiabatic couplings in the avoided crossing regime, and inelastic scattering. The former probes the charge density and delivers direct information on the evolving molecular geometry. The latter two contributions are weaker and carry spatial information of the transition charge densities (off-diagonal elements of the charge-density operator). Simulations are presented for the nonadiabatic harpooning process in the excited states of sodium fluoride.
I Introduction
X-ray diffraction Guinier (1994); Als-Nielsen and McMorrow (2011); Thibault and Elser (2010) has been used for over a century to probe the structure of crystals and has been extended to diffuse scattering from liquids, probing nearest-neighbor distances, and serves as inspiration for the conceptually similar electron diffraction technique Ben-Nun et al. (1996); Siwick et al. (2003). Time-resolved X-ray diffraction (TRXD) can track the structural changes that characterize phase transitions and chemical reactions and has been actively pursued to create movies of elementary molecular events Bratos et al. (2002); Siwick et al. (2003); Coppens et al. (2005); Ihee et al. (2005); Wulff et al. (2006); Cammarata et al. (2008); Siders et al. (1999); Woerner et al. (2010); Coppens (2011); Neutze and Moffat (2012); Yang et al. (2016); Larsson, Falcone et al. (1998); Glownia et al. (2016); Kochise Bennett and Mukamel . Free electron lasers allow extremely bright and ultrafast X-ray pulses. These should make it possible to push diffraction to the single-molecule limit, Stevens (2000); McPherson (1999); Hajdu (2000); Chapman (2009); Starodub et al. (2012); Kahra et al. (2012) eliminating the need for time-consuming crystal preparation. In addition, their femtosecond timescale opens up the possibility of tracking attosecond electronic dynamics while the brightness may permit even weak signals, such as inelastic scattering from transient electronic coherences, to be measured Altarelli et al. (2006); Feldhaus, Arthur, and Hastings (2005); McNeil and Thompson (2010); Chapman et al. (2011); Bostedt et al. (2013); Barty, Küpper, and Chapman (2013).
In this paper, we show how TRXD may be used to obtain real-time stroboscopic snapshots of nonadiabatic molecular dynamics. Nonadiabatic processes control virtually all photochemical and photophysical processes in molecules. For a single vibrational coordinate, this results in avoided crossings. With two or more vibrational degrees of freedom, conical intersections (CoIns) become possible. As a molecule passes through a conical intersection Domcke, Yarkony, and Köppel (2011) or avoided crossing, a short-lived electronic coherence is created, which can be spectroscopically detected Kowalewski et al. (2015, 2016) by X-rays. Examples for a photochemical reactions that are mediated by a CoIn and has been studied by TRXD Minitti et al. (2015) is the ring-opening reaction in cyclohexadiene Hofmann and de Vivie-Riedle (2001); Tamura et al. (2006) and the cis/trans isomierization in the photoactive yellow protein Pande et al. (2016). Potential signatures in TRXD signals might also be useful to measure the geometric (Berry) phase Xiao, Chang, and Niu (2010), which has so far eluded detection in molecules.
We examine the elastic and inelastic contributions to the diffraction pattern that stem from the coupled nonadiabatic electronic+nuclear dynamics in the vicinity of an avoided crossing. Time-resolved scattering from photoexcited molecules in the gas phase is given by an incoherent sum of single-molecule contributions, contains elastic and inelastic terms, and may depend on electronic coherence Cao and Wilson (1998); Henriksen and Møller (2008). We calculate the TRXD by an ensemble of molecules prepared in a superposition of valence electronic and vibrational states. We identify five distinct contributions to the signal and study their relative intensity and time-resolved diffraction pattern. Contributions from electronic coherences, which are created in the avoided crossing region are of particular interest. The underlying molecular quantities are the transition charge densities between electronic states. The nonadiabatic dynamics of sodium iodide has been investigated in Ref. Lorenz, Møller, and Henriksen which did not address the signatures of electronic coherences. Dixit et al. Dixit, Vendrell, and Santra (2012) investigated diffraction of electronic superpositions but did not include nuclear dynamics. We examine the nonadiabatic coupled electronic+nuclear motions and the signatures of electronic coherences in the diffraction signal of a similar molecule, sodium fluoride.
II The Interplay of Populations and Coherences in Single-Molecule Diffraction of Nonadiabatic Dynamics
Our study starts with the following expression for the off-resonant scattering signal in the gas phase (see appendix A for derivation).
[TABLE]
where is the temporal envelope of the X-ray pulse, stands for expectation value over the nuclear and electronic states, and is the spatial Fourier transform of the charge-density operator. Note that Eq. (1) comes with while the classical equation for diffraction in crystals comes with .
The total charge-density operator for a system composed of molecules can be written as a sum of the charge densities from each molecule
[TABLE]
where is the center of molecule . This separation is exact for a sufficiently dilute system such that the molecules have non-overlapping charge distributions, since each electron (the fundamental X-ray scatterer) can be rigorously assigned to a specific molecule. The elastic diffraction signal from a system initially in the ground state is
[TABLE]
where is the ground-state charge density in -space and is the scattering momentum transfer. For identical molecules, the charge-density matrix elements of each molecule only differ by the spatial phase factor associated with the location of the molecule and we may drop the subscript on .
We now apply these results to a molecular model consisting of two electronic states and a single active nuclear coordinate (Fig. 1). The time-dependent wavefunction of each molecule in the ensemble will be expanded in the adiabatic basis
[TABLE]
where is the (normalized) nuclear wave packet on the adiabatic electronic state and are the electronic state amplitudes. The time evolution of is governed by the field-free nuclear Hamiltonian , which includes the nonadiabatic coupling matrix elements to account for CoIns or avoided crossings in the time evolution. The elements of the reduced electronic density matrix are given by , which depends on the dephasing caused by the nuclear wave packet overlap in states and . Expanding the time-dependent densities in the electronic states using the diagram in Fig. 1 results in
[TABLE]
Fig. 2 gives the complete set of diagrams. For a two electronic state model the diagrams in Fig. 2 result in the following five contributions to the signal:
[TABLE]
where the electronic populations and coherences are given by the diagonal and off-diagonal elements of the density matrix respectively and we have defined the electronic-state matrix elements of the charge-density operator (which remains an operator in the nuclear space and we ommit the dependence for brevity). Equation (6) agrees with earlier results Cao and Wilson (1998); Henriksen and Møller (2008) but identifies the different contributions in the adiabatic basis.
The first two terms on the right-hand side of Eq. (6) (i) and (ii) represent the elastic diffraction from states and respectively, which encode the time evolution of the nuclear wave packets in the two electronic states. The next two terms, (iii) and (iv), represent the inelastic () scattering from the electronic ground and excited state populations. The last term (v) is due to scattering off electronic coherences between and .
Diffraction is often analyzed by assuming that the molecular electronic charge density is solely composed from the atomic densities. In case the molecule is in the electronic state and Eq. (6) can be simplified by the independent atom approximation Bartell and Gavin (1964); Waller and Hartree (1929); Yang et al. (2016):
[TABLE]
where is the atomic charge density of the th atom in the molecule and is its phase factor. This widely used expression approximates term (i) in Eq. (6) but does not account for inelastic scattering events and contributions due to electronic coherences. Our theory explicitly seperates inelasticities, which are described by transition charge densities () that interfere with ground and excited state terms .
III Avoided Crossing Dynamics in Sodium Fluoride
We now present and discuss the five contributions to the diffraction signal from sodium fluoride. This molecule possesses a similar electronic structure to sodium iodide, the avoided crossing of which was studied in Zewail’s landmark optical experiment Rose, Rosker, and Zewail (1989). Excited-state diffraction of sodium iodide has been calculated Lorenz, Møller, and Henriksen by including the nonadiabatic dynamics but focusing solely on the elastic scattering processes (corresponding to terms (i) and (ii)). An avoided crossing between the ionic and covalent state at 8 Å, known as harpooning, creates an electronic coherence in the course of the time evolution of the excited state nuclear wave packet (see Fig. 3).
Iodine is a strong X-ray scatterer and its large nuclear charge leads to a charge density distribution which is heavily dominated by its core electrons. While this is still the case for molecular form factors of lighter element compounds, they have a more prominent contribution from valence electrons compared to the core electrons. The coherence contributions, which depend on the transition densities and are dominated by the rearrangement of valence electrons are thus expected to be relatively stronger in sodium fluoride than in sodium iodide.
III.1 Electronic Structure Calculations and Nonadiabatic Wave packet dynamics
The electronic structure of NaF was calculated with the program package Molpro Werner et al. (2015) at the CAS(8/9)/MRCI/aug-cc-pVTZ level of theory. A Douglas-Kroll-Hess 10th-order correction has been used Douglas and Kroll (1974); Hess (1986) to account for relativistic effects caused by the core electrons. All densities were evaluated from the state specific charge density matrices (and transition charge density matrices) , expanded in the atomic orbital basis functions :
[TABLE]
Both the transition dipole and the integrated transition density, shown in Fig. 4, peak at the avoided crossing point.
The matrix elements of the electronic density operator are displayed in Fig. 5. For clarity, only the projection along the direction of molecular axis obtained by integrating over the perpendicular directions is shown. The diagonal density (Fig. 5 (a)) is clearly dominated by contributions from the core electrons and the stripe pattern reflects the bond length in reciprocal space (see Eq. (1)). The transition density (Fig. 5 (b)) mainly contains contributions from the valence orbitals involved in the transition. It is about 4 orders weaker than the diagonal matrix element (Fig. 5 (a)). However, it peaks at the avoided crossing, making it most suitable for the detection of inelastic contributions. The mixed matrix element (Fig. 5 (c)) is a product of the nuclear densities and the transition densities.
Nuclear wave packet dynamics simulations were carried out on a numerical grid with 1200 grid points for the nuclear coordinate (extending from 2 to 24 Å) and the electronic states and . The Hamiltonian, which describes the coupled electronic and vibrational degrees of freedom, is given by:
[TABLE]
where
[TABLE]
is the kinetic operator of the nuclei, the reduced mass of the nuclei, and
[TABLE]
approximates the non-adiabatic couplings Domcke, Yarkony, and Köppel (2011); Hofmann and de Vivie-Riedle (2001).
[TABLE]
is the non-adiabatic coupling matrix element between and and has been obtained by a fit to values calculated with the DDR routine in MOLPRO Werner et al. (2015). The fitted parameters are , , and (all values in atomic units).
We assume a Gaussian pump-pulse envelope:
[TABLE]
where is the full width at half maximum of the intensity profile . The probe-pulse is not included in the propagation but is treated pertubatively and included in the final signal calculation (Eq. (1)). The wave function is obtained by propagating the vibrational ground state of the state with a Chebychev scheme Ezer and Kosloff (1984) using the Hamiltonian Eq. (9). The kinetic operator is modified with a perfectly matched layer Nissen, Karlsson, and Kreiss (2010) for to avoid spurious reflections at the edges of the grid ( Å). The signal is then obtained by evaluating Eqs. (3) and (5) and inserting the time-dependent wave functions and density operators (, as shown in Fig. 5). We use the adiabatic basis but the calculation is exact. The electronic coherence is obtained from the combined electronic-nuclear wave function as the overlap of the nuclear wave packets:
[TABLE]
This results in the decay and revival of the electronic coherence.
The wave packet dynamics in the excited state potential () is depicted in Fig. 6. It passes through the avoided crossing between 200 and 240 fs and reaches its outer turning point around 500 fs. The time-dependent excited state population alongside with the magnitude of the electronic coherence is shown in Fig. 7.
The two relevant valence states (Fig. 3) are the X ground state and the A state (referred to as and in the following). A UV pump-pulse creates an excited-state population %, triggering the nuclear wave packet dynamics in states and that is subsequently probed with a fs X-ray probe pulse. The time dependent excited-state population and the coherence are displayed in Fig. 7. At around 200 fs, the excited-state nuclear wavepacket first reaches the avoided crossing and returns to the crossing between 750 and 900 fs.
IV The Diffraction Signal
Figure 8 shows the diffraction pattern as well as the relative magnitude of the five contributions to the signal in Eqs. (6) and (1).
The contributions to the diffraction signal are shown as labeled in Eq. (6) ((i) through (v), corresponding to the diagrams of Fig. 2). The elastic diffraction signal, which stems from the charge density , is shown in Fig. 8(i). The time evolution represents the wave packet motion, i.e., the fringe spacing increases as the wave packet moves towards a longer bond length. The actinic pump-pulse (full width at half maximum 10 fs) also creates a non-stationary nuclear wave packet in the electronic ground state. This ground-state hole has comparable magnitude to the excited state contribution Fig. 8(i). Figure 8(ii) shows the diffraction signal from the ground state density. The interference fringes are signatures of an oscillating vibrational wave packet in the ground-state potential. This hole burning phenomena will occur for pump-pulses that have bandwidths smaller than the bandwidth of the Franck-Condon region.
The inelastic contribution that stems solely from the transition densities and the excited-state wavepacket in Fig. 8(iii), is 4 orders of magnitude weaker. It carries no information about the electronic coherence but is dominated by the shape and magnitude of the transition density and is closely related to the transition dipole moment. This contribution varies widely over time since the nuclear wavepacket enters a region, where the transition dipole vanishes. The inelastic scattering from the ground state shown in Fig. 8(iv), is also modulated by the wavepacket motion. Compared to Fig. 8(iii), its intensity is only weakly modulated since it never reaches a region where the transition dipole moment vanishes.
Figure 8(v) depicts the combined contribution of inelastic scattering of the electronic coherences. This contribution is responsible for the time-evolving density caused by the electron dynamics Dixit, Vendrell, and Santra (2012). At 220 fs, when the wavepacket hits the avoided crossing regime, an electronic coherence is created, resulting in a slow temporal oscillation that spreads over a wide range in -space. This contribution is 3 orders of magnitude weaker than the excited-state density (Fig. 8(i)) but one order of magnitude larger than the other inelastic contributions (iii) and (iv). Another contribution appears at around 800 fs, which stems from the returning wavepacket but it is much weaker due to the larger spread of the wavepacket. When the wavepacket returns to the Franck-Condon point, a larger spike in the coherence is visible in Fig. 7 at around 1100 fs. This contribution is averaged out in Fig. 8(v) due to the probe-pulse length and would require an attosecond rather than a femtosecond pulse to observe.
Figure 9 shows the coherence contribution in Fig. 8(v) in real-space (Fourier transform). The first passage through the avoided crossing at 200 fs shows a clear signature at 8 Å, thus giving a hint of where the electronic coherence has been created. The second passage at around 800 fs is also visible.
V Conclusions
In conclusion, the simulated gas-phase or single-molecule diffraction signal of sodium fluoride undergoing nonadiabatic avoided crossing dynamics contains signatures of the created electronic coherence on top of the dominant ground- and excited-state wavepacket motion. The diffraction signal depends on the ground- and the excited-state charge densities as well as the transition charge density that causes the inelastic contribution (v). These densities depend on time through the interatomic distance, which can be extracted directly from the diffraction signal. The shape of the nuclear wavepacket can be qualitatively retrieved without further phase reconstruction. For diatomic molecules, this allows to create a molecular movie out of the diffraction data. The coherence contributions do not merely indicate that a coherence has been created but also reveal where it has been created. Its contribution is significantly weaker than elastic scattering processes and appears as a rapid oscillation on top of the diffraction pattern. It will be interesting to explore other nonlinear optical signals where the coherence contribution is more pronounced and possibly background free Kowalewski et al. (2015, 2016). Finally, we note that, by including additional nuclear coordinates our approach may be used to predict signatures of CoIns in polyatomic molecules. Extended nonlinear probe schemes may be capable of directly imaging the transition density.
Acknowledgements.
The support of the Chemical Sciences, Geosciences, and Biosciences division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy through award No. DE-FG02-04ER15571 is gratefully acknowledged. Support for K.B. was provided by DOE. M.K. gratefully acknowledges support from the Alexander von Humboldt foundation through the Feodor Lynen program.
Appendix A The Scattering Signal
We start with the basic definition of the signal derived from time-dependent perturbation theory, which allows its convenient dissection into its different contributions. In a previous workBennett et al. (2014), we derived the following expressions for single-molecule frequency-resolved diffraction signals
[TABLE]
In Eq. (15), is the vector potential envelope for the X-ray probe, is a frequency gating (detector sensitivity) function, stands for the set of parameters defining the X-ray field, and is the momentum transfer ( is the direction of the scattered wavevector). In standard applications, the molecules that compose the sample are assumed to have identical charge distributions and the subscripts on the charge density should be dropped, as we will do for the remainder of this manuscript. Assuming no frequency resolution, we have for the frequency gating function. The long-range (inter-molecular) structure of the sample is captured by the structure factors
[TABLE]
in terms of which the diffraction signals can be written as
[TABLE]
where we have substituted the electric field envelopes for the vector potential. For near-elastic scattering, we approximate , which is nearly valid even for inelasticities of several eV since the central frequency of the X-ray pulse is on the order of 10keV. Similarly, the momentum transfer is approximated as independent of frequency. For the purposes of time-resolved diffraction studies, a time-domain expression is more convenient to simulate due to the nuclear motion. We thus substitute the time-domain charge density operator
[TABLE]
where we work in the interaction picture so that the operator time-dependence is through the field-free propagator, to obtain
[TABLE]
where we have replaced by in the argument in accordance with the quasi-elastic approximation. Upon carrying out the integration and using , finally results in Eq. (1)
Appendix B The Electronic Charge Density Operator
In this section, we discuss the operator nature of the charge density and its consequences. In this section, we will ignore nuclear dependence and will begin by considering a one-electron system. We seek an operator such that the expectation value in a given state is the charge density
[TABLE]
This identifies the real-space matrix elements of the electronic charge density field operator
[TABLE]
For a state decomposed into eigenmodes , we have
[TABLE]
where are the electronic populations and coherences. Note that this matches the usual field-theoretic definition of the charge density
B.1 The One-Electron Charge Density Operator of a Many-Electron System
In this section, we extend the reasoning of the previous section to an -electron state . The real-space identity operator in the space spanned by such states is
[TABLE]
and the one-electron charge density is Szabo and Ostlund (1996)
[TABLE]
Since the charge-density operator is a one-electron operator, we have the straightforward -electron generalization of Eq. (21)
[TABLE]
which is directly confirmed by substitution into Eq. (24) and gives
[TABLE]
where we have identified
[TABLE]
We note that this result can equally well be obtained by use of real-space field operators for many-electron systems as explicated by Cederbaum Kuleff and Cederbaum (2011); Cederbaum (2016). Moreover, Eq. (27) is readily generalized to account for nuclear degrees of freedom as
[TABLE]
where the circumflex indicates that the left hand side remains an operator in the nuclear subspace due to dependence on
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Guinier (1994) A. Guinier, X-ray diffraction: in crystals, imperfect crystals, and amorphous bodies (Courier Dover Publications, 1994).
- 2Als-Nielsen and Mc Morrow (2011) J. Als-Nielsen and D. Mc Morrow, Elements of modern X-ray physics (Wiley, Hoboken, 2011).
- 3Thibault and Elser (2010) P. Thibault and V. Elser, “X-ray diffraction microscopy,” Annu. Rev. Cond. Mat. Phys. 1 , 237–255 (2010).
- 4Ben-Nun et al. (1996) M. Ben-Nun, T. J. Martínez, P. M. Weber, and K. R. Wilson, “Direct imaging of excited electronic states using diffraction techniques: theoretical considerations,” Chem. Phys. Lett. 262 , 405–414 (1996).
- 5Siwick et al. (2003) B. J. Siwick, J. R. Dwyer, R. E. Jordan, and R. D. Miller, “An atomic-level view of melting using femtosecond electron diffraction,” Science 302 , 1382–1385 (2003).
- 6Bratos et al. (2002) S. Bratos, F. Mirloup, R. Vuilleumier, and M. Wulff, “Time-resolved x-ray diffraction: statistical theory and its application to the photo-physics of molecular iodine,” J. Chem. Phys. 116 , 10615–10625 (2002).
- 7Coppens et al. (2005) P. Coppens, I. I. Vorontsov, T. Graber, M. Gembicky, and A. Y. Kovalevsky, “The structure of short-lived excited states of molecular complexes by time-resolved x-ray diffraction,” Acta Crystallographica Section A: Foundations of Crystallography 61 , 162–172 (2005).
- 8Ihee et al. (2005) H. Ihee, M. Lorenc, T. K. Kim, Q. Y. Kong, M. Cammarata, J. H. Lee, S. Bratos, and M. Wulff, “Ultrafast x-ray diffraction of transient molecular structures in solution,” Science 309 , 1223–1227 (2005).