Infrared spectroscopy of molecular ions in selected rotational and spin-orbit states
Ugo Jacovella, Josef A. Agner, Hansj\"urg Schmutz, Johannes Deiglmayr,, Fr\'ed\'eric Merkt

TL;DR
This paper introduces a novel experimental setup for recording infrared spectra of molecular ions in specific rotational and spin-orbit states, enabling detailed spectroscopic analysis of selected ion states.
Contribution
The work presents a new state-selective IR spectroscopy method for molecular ions using mass-analyzed-threshold-ionization and reaction detection, improving state specificity over previous techniques.
Findings
IR spectra of C₂H₂⁺ in specific rotational levels obtained
Method demonstrates selective excitation and detection of ion states
Spectra reveal details of spin-orbit components in the ground state
Abstract
First results are presented obtained with an experimental setup developed to record IR spectra of rotationally state-selected ions. The method we use is a state-selective version of a method developed by S. Schlemmer, D. Gerlich and coworkers (Int. J. Mass. Spec. 185, 589 (1999); J. Chem. Phys. 117, 2068 (2002)) to record IR spectra of ions. Ions are produced in specific rotational levels using mass-analyzed-threshold-ionization spectroscopy. The state-selected ions generated by pulsed-field ionization of Rydberg states of high principal quantum number () are extracted toward an octupole ion guide containing a neutral target gas. Prior to entering the octupole, the ions are excited by an IR laser. The target gas is chosen so that only excited ions react to form product ions. These product ions are detected mass selectively as a function of the IR laser wavenumber. To…
Click any figure to enlarge with its caption.
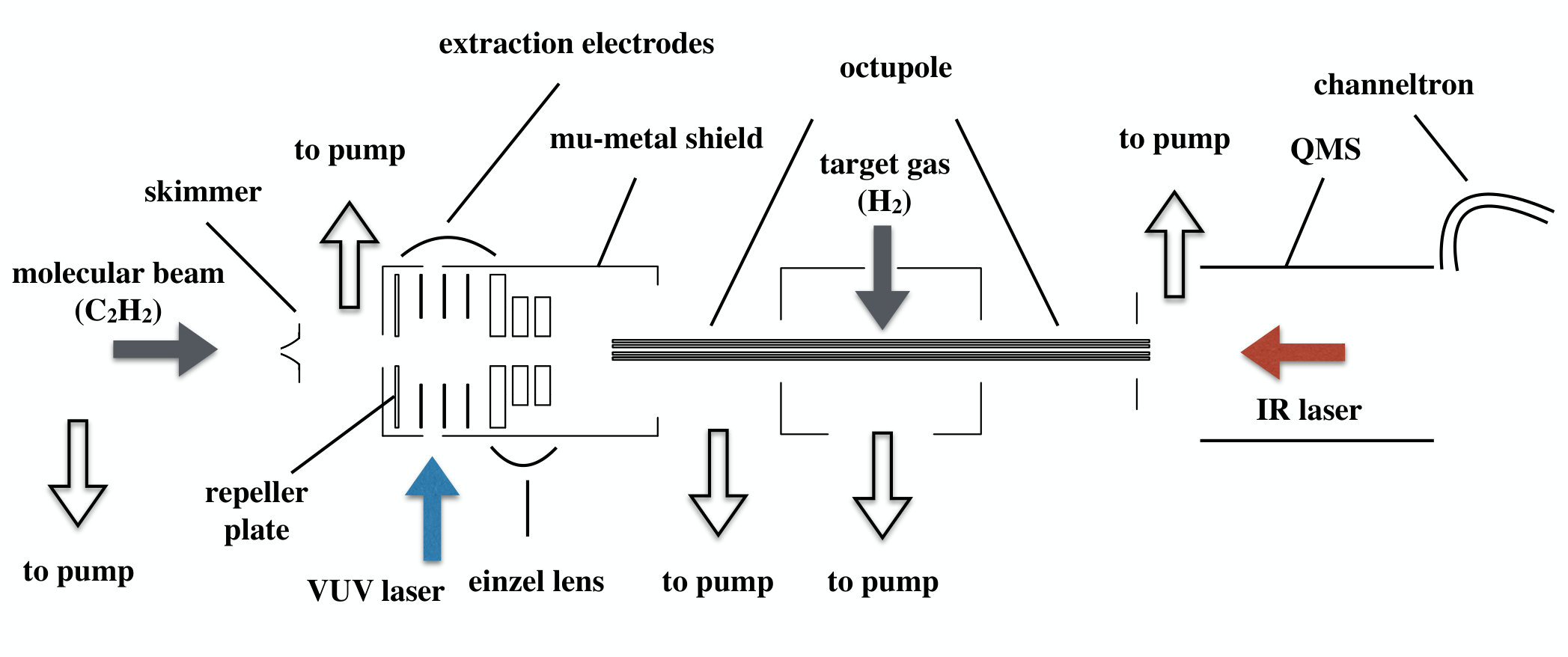 Figure 1
Figure 1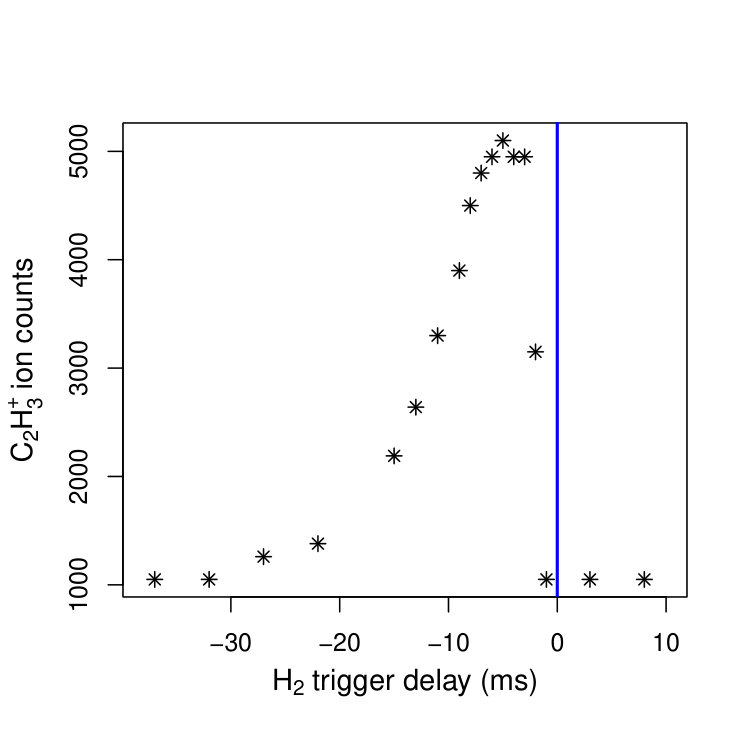 Figure 2
Figure 2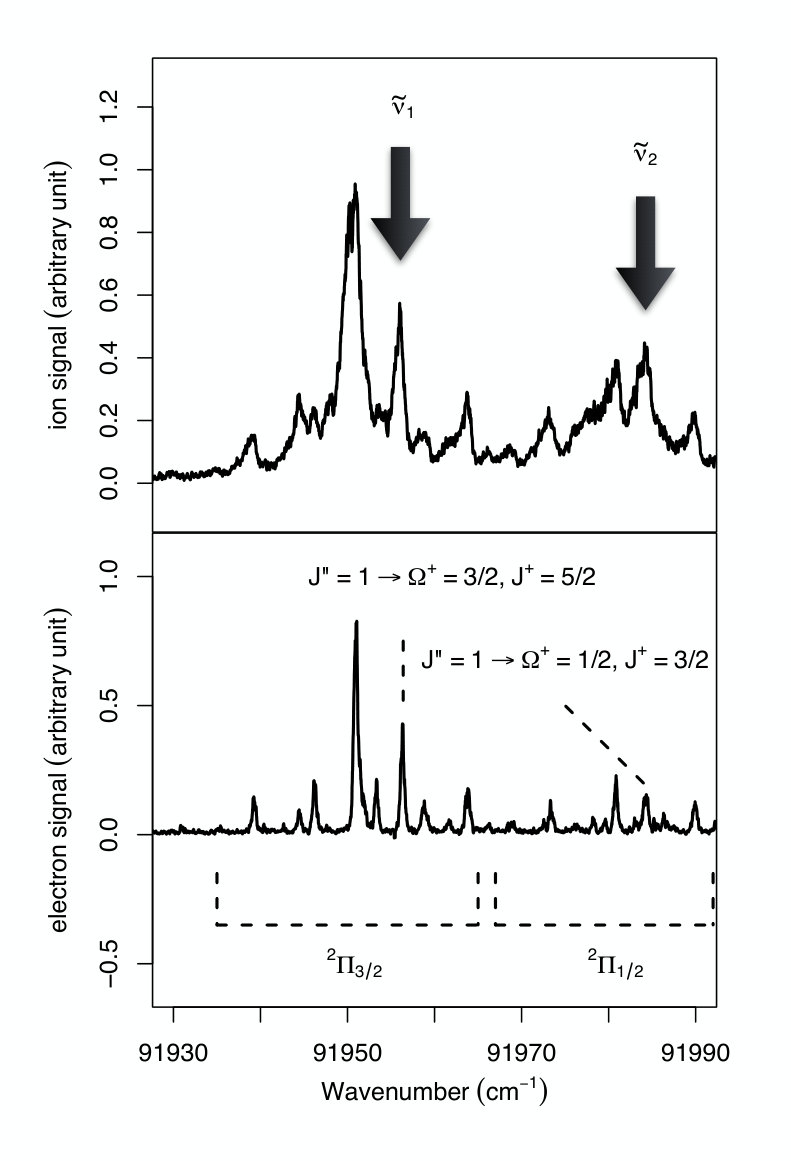 Figure 3
Figure 3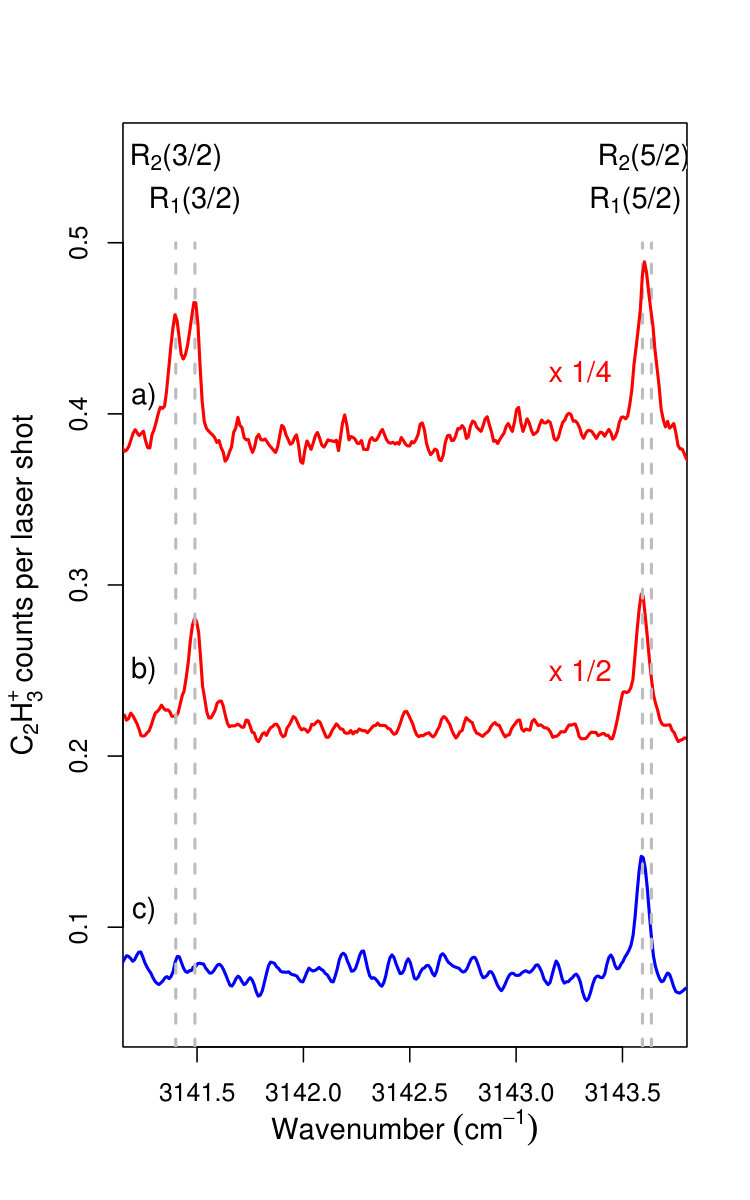 Figure 4
Figure 4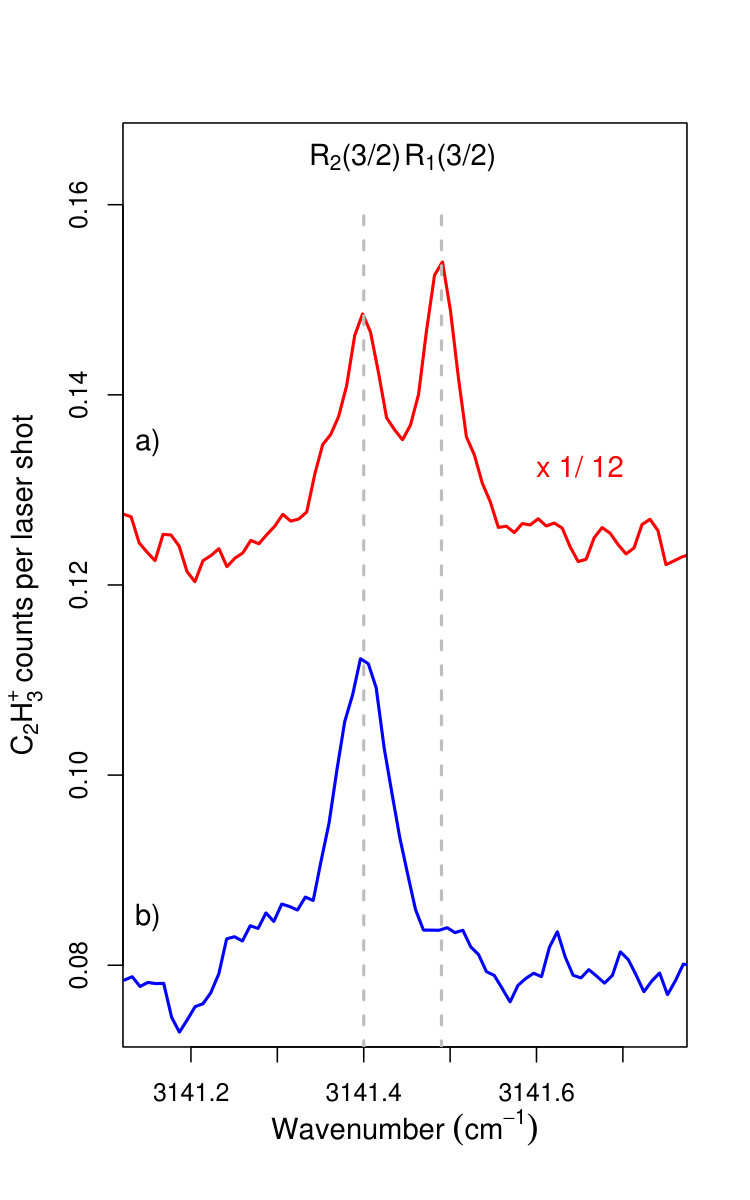 Figure 5
Figure 5| This work | Ref. [Jagod1992, ] | |
|---|---|---|
| R1(3/2) | 3141.488(20) | 3141.491 |
| R2(3/2) | 3141.400(20) | 3141.402 |
| R1(5/2) | 3143.597(20) | 3143.594 |
| R2(5/2) | 3143.629(30) | 3143.636 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Infrared spectroscopy of molecular ions in selected rotational and spin-orbit states
U. Jacovella
Laboratorium für Physikalische Chemie, ETH Zürich, CH-8093 Zurich, Switzerland
J. A. Agner
Laboratorium für Physikalische Chemie, ETH Zürich, CH-8093 Zurich, Switzerland
H. Schmutz
Laboratorium für Physikalische Chemie, ETH Zürich, CH-8093 Zurich, Switzerland
J. Deiglmayr
Laboratorium für Physikalische Chemie, ETH Zürich, CH-8093 Zurich, Switzerland
F. Merkt
Laboratorium für Physikalische Chemie, ETH Zürich, CH-8093 Zurich, Switzerland
Abstract
First results are presented obtained with an experimental setup developed to record IR spectra of rotationally state-selected ions. The method we use is a state-selective version of a method developed by S. Schlemmer, D. Gerlich and coworkers (Int. J. Mass. Spec. 185, 589 (1999); J. Chem. Phys. 117, 2068 (2002)) to record IR spectra of ions. Ions are produced in specific rotational levels using mass-analyzed-threshold-ionization spectroscopy. The state-selected ions generated by pulsed-field ionization of Rydberg states of high principal quantum number () are extracted toward an octupole ion guide containing a neutral target gas. Prior to entering the octupole, the ions are excited by an IR laser. The target gas is chosen so that only excited ions react to form product ions. These product ions are detected mass selectively as a function of the IR laser wavenumber. To illustrate this method, we present IR spectra of C2H in selected rotational levels of the and spin-orbit components of the vibronic ground state.
Suggested keywords
pacs:
Valid PACS appear here
††preprint: AIP/123-QED
I Introduction
Many important molecular cations such as H, CH, CH, C2H, C2H, C2H, C5H, C6H, O, etc., have complex or unknown spectra. The presence of unpaired electrons, orbital degeneracy andor large-amplitude motions in these ions leads to intricate energy-level structures and congested spectra. Once measured, the spectra can resist assignments for years, if not for decades. Striking examples include the Carrington-Buttenshaw-Kennedy spectrum of H,Carrington1982 the White-Tang-Oka spectrum of CH,White1999 and the spectrum of CH.signorell1999 ; woerner2009
The difficulties in studying cations arise from their high reactivity and the fact that space-charge effects limit their densities in the gas phase. In most cases, ions are generated in hot environments so that the population is distributed over many quantum states, which further reduces the sensitivity of spectroscopic experiments and complicates spectral assignments.
Several approaches have been developed to overcome these difficulties. These can be classified into three categories: i) the direct measurement of ion spectra by absorption or emission spectroscopy, Herzberg1971 ; Saykally1981 ; Hirota1992 ; Hirota2000 the sensitivity of which is greatly enhanced by exploiting modulation techniques,Oka2003 ; Stephenson2005 ; Hodges2013 ii) the indirect measurement of ion spectra by photoelectron spectroscopy of the neutral parent molecules, for instance by pulsed-field-ionization zero-kinetic-energy (PFI-ZEKE) photoelectron spectroscopy, Muller1991 ; Merkt2011 and iii) the measurement of spectra by detecting photoinduced processes, e.g. dissociation products,Dzhonson2006 ; Rizzo2015 reaction products,Schlemmer1999 ; Schlemmer2002 the suppression of complex formation,Duncan2003 ; Chakrabarty2013 or specific processes in ion traps resulting from photoexcitation Tong2011 . A variant of iii) consists of recording the spectra of weakly bound complexes of the ion of interest with rare gas atoms and monitoring the rare-gas-atom loss.Duncan2011 Significant advances could be made in this last category with the introduction of 22-pole ion traps in combination with buffer-gas cooling Gerlich1993 and the use of sympathetically cooled molecular ions in laser-cooled Coulomb crystals.Willitsch2012 Several recent review articles provide an overview of this very active field.Willitsch2011 ; Amano2011
We present here a method for recording spectra of state-selected ions and illustrate it with the example of a measurement of the asymmetric-stretch fundamental band (3) of C2H. The transitions are detected by monitoring the C2H product of the reaction
[TABLE]
which is not observable for C2H ions in their vibronic ground state, as was already exploited in the pioneering experiment of Schlemmer Schlemmer2002 . In Eq. (1), is the total-angular-momentum quantum number excluding nuclear spin and is the quantum number associated with the projection of on the molecular axis. C2H is particularly well suited to test the performance of our method because the transition frequencies are accurately known from the work of Oka and coworkers Jagod1992 and the spin-orbit and rotational structure of the photoelectron spectrum of the X+ X ionizing transition has been fully resolved by PFI-ZEKE photoelectron spectroscopy Rupper2004 .
Carrying out the experiments with state-selected ions makes the spectral assignments straightforward, reduces spectral congestion and allows in principle the measurement of infrared spectra of vibrationally excited ions.
II Experimental procedure
The first step of our experimental procedure consists in preparing the C2H cations in selected rotational levels of the ground electronic state and generating a beam of these ions. This step is accomplished using mass-analyzed-threshold-ionization (MATI) Zhu1991 ; Merkt1993 combined with single-photon excitation of a cold sample of C2H2 molecules in a supersonic beam using narrow-band vacuum-ultraviolet (VUV) laser radiation.
The C2H ions are irradiated with an IR laser pulse before being extracted toward an octupole ion guide containing the neutral H2 target gas. The IR transitions are detected by monitoring the C2H product-ion yield as a function of the IR-laser wavenumber. The experimental setup is represented schematically in Fig. 1.
The pulsed tunable VUV laser radiation (repetition rate 25 Hz, pulse duration 2 ns) was generated by resonance-enhanced difference-frequency mixing in a pulsed beam of xenon Rupper2004 using the (5p)6 (5p)56p[1/2]0 two-photon resonance of Xe at cm*-1* and two commercial single-grating dye lasers and a -barium-borate doubling crystal.
The pulsed tunable IR laser radiation was generated by difference-frequency mixing in a KTiOAsO4 (KTA) crystal. The KTA was cut at angles of , and had a dimension of mm3. The seeded 532 nm output of the Nd:YAG laser and the output of a double-grating tunable dye laser operated in the range 638-642 nm were overlapped using a dichroic mirror and directed into the crystal. To separate the generated IR laser beam from the input laser beams, these were aligned so as to intersect in the KTA crystal at a small angle (). A slit was used to block the 532 nm and 640 nm laser beams but transmit the IR laser beam before the IR beam entered the experimental chamber through a CaF2 window. The IR radiation had a measured bandwidth of 0.035 cm*-1* and a pulse energy of 200 J. Its wavenumber was calibrated at an absolute accuracy of 0.02 cm*-1* by measuring the wavenumber of the 532 nm and 640 nm lasers with a wavelength meter.
A pulsed valve was employed to form a supersonic beam of a mixture of 20 acetylene and 80 argon (volume ). The beam was skimmed prior to entering the photoionization chamber, where it intersected the VUV laser beam at right angles on the axis of a set of five parallel cylindrical extraction plates.Rupper2004
The ions were state selected using two successive electric-field pulses,Merkt1993 a discrimination pulse of +690 mV/cm, which removed all prompt ions produced by the VUV laser, followed by a field-ionization and ion-extraction pulse of 5.5 V/cm, generated by applying a pulsed electric potential of +32 V on the repeller plate (see Fig. 1) of the extraction region. The IR excitation was carried out either during the discrimination pulse or in the field-free interval between discrimination and extraction pulses, or immediately following field ionization with the extraction pulse. In this way the Doppler shift of the IR transition only originated from the velocity of the supersonic beam (600 m/s), and was 0.006 cm*-1* at 3000 cm*-1*, i.e., more than 5 times less than the IR-laser bandwidth. The IR spectra turned out not to be sensitive to the exact timing of the IR laser pulse as long as the IR excitation took place before significant acceleration of C2H by the extraction pulse. We therefore conclude that the presence of the Rydberg electron has no effect on IR transition frequencies within the precision limit of our experiment and of electric fields up to 5.5 V/cm. However, we observed a slight broadening (from 0.035 cm*-1* to 0.050 cm*-1*) of the IR transitions recorded before the field-ionization pulse (see below).
The ions extracted toward the reaction zone were produced at a potential of V, determined by the applied potential and the experimental geometry, and were slowed down on their way to the octupole containing the target gas H2 by applying a V bias potential to the octupole rods. The exact value of the bias potential was carefully adjusted so that i) the kinetic energy of the ions entering the reaction zone was too low for a reaction to occur without prior excitation of C2H with the IR laser, and ii) the prompt ions were all rejected.
The target gas H2 was introduced from the side using a pulsed valve with orifice located 10 cm away from the octupole axis. Rather than monitoring the product ions resulting from the reaction of C2H with H2 molecules in the supersonic beam, we found it more efficient to open the H2 valve early and to let the H2 molecules thermalize to 300 K prior to the arrival of the C2H beam in the octupole. The optimal valve-opening time was determined by monitoring the C2H product-ion yield, as illustrated in Fig. 2. In this figure, the vertical line indicates the temporal position of the VUV laser pulse and the stars represent the C2H ion signal. The maximum yield was obtained by opening the H2 valve about 6 ms before the VUV laser pulse. After the reaction, the product ions (C2H) were guided toward a commercial quadrupole mass spectrometer coupled with an off-axis channeltron, where they were detected mass-selectively.
III Results
All assignments are presented using Hund’s case (a) nomenclature, which is adequate for low values of . The IR transitions are labeled R1,2(), where R is the standard notation for transitions associated with =+1, is the rotational quantum number of the initial state and the subscripts 1 and 2 refer to the F1 () and F2 () spin-orbit components of the X+ ground electronic state of C2H.
Figure 3 compares the MATI spectrum of the X+ (=0) X (=0) ionizing transition of C2H2 (upper trace) with the high-resolution PFI-ZEKE photoelectron spectrum of the same transition reported by Rupper Rupper2004 . Several lines of the MATI spectrum correspond to single transitions, such as the X+ ( = 5/2) X () transition at the position cm*-1* (arrow marked in Fig. 3). At this spectral position, the pulse sequence used to form the ion beam guarantees that the ions in the beam are in the X+ () level prior to IR excitation. At the position cm*-1* (arrow marked in Fig. 3), the C2H ions are produced in the rotational level of the spin-orbit component. By turning off the discrimination pulse, one can also generate an ion beam to which all ions contribute that can be generated by direct photoionization at the selected wavenumber.
These advantages are illustrated in Fig. 4. Trace a) was measured without discrimination pulse with the VUV wavenumber set at 92080 cm*-1*, , well above the X+ X band. At this position all optically accessible rotational levels of the X+ and spin-orbit components are generated. The spectral region displayed in this spectrum consists of R transitions from the and 5/2 levels of both spin-orbit components. When the VUV laser wavenumber is set at 91954 cm*-1*, , below the onset of the X+ X band in the MATI spectrum (see Fig. 3), but still without discrimination pulse, the two R2 lines disappear as can be seen in the middle trace (trace b)) in Fig. 4. The R1(5/2) line is the only one remaining when the VUV laser is tuned to the position of in Fig. 3 and the discrimination pulse is switched on (trace c) in Fig. 4). This trace thus demonstrates full rotational state selectivity in the X+ spin-orbit component.
Full rotational state selectivity can also be achieved in the upper () spin-orbit component, as illustrated in Fig. 5. This figure compares the IR spectrum near the R1,2(3/2) line pairs recorded with the VUV laser wavenumber fixed at the position marked in Fig. 3 with (lower trace) and without (upper trace) discrimination pulse. This state selectivity is possible because the H2 gas density in the reaction zone is low enough to ensure single-collision conditions and thus to avoid state redistribution by inelastic collisions prior to the reaction.
The lines observed in the IR spectrum presented in Figs. 4 and 5 have a full width at half maximum of 0.035 cm*-1*, limited by the bandwidth of the IR laser. In IR spectra recorded by applying the IR laser pulse before the field ionization pulse, we did not observe any frequency shift of the IR transitions but a slight broadening, which we attribute to the presence of the high- Rydberg electron. The transition wavenumbers, corrected for the estimated Doppler shift of 0.006 cm*-1*, are compared in Table 1 to the more precise transition wavenumbers reported in Ref. [Jagod1992, ] with which they are in excellent agreement.
IV Conclusion
We developed a method and built a new apparatus to record infrared spectra of mass- and state-selected cations. The method relies on the preparation of the individual vibrational and rotational state of molecular cations by photoionization and field-ionization methods and on the detection of the product of reactions induced by the absorption of infrared radiation. The state selectivity of the method reduces spectral congestion, which facilitates assignments. The main obstacle to the general use of this method is the necessity to identify a reaction with the appropriate kinetics and thermodynamical properties.
Acknowledgements.
This work is supported financially by the Swiss National Science Foundation under project Nr. 200020-159848.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1(1) A. Carrington, J. Buttenshaw and R. Kennedy, Mol. Phys. 45 , 753 (1982).
- 2(2) E. T. White, J. Tang and T. Oka, Science 284 , 135 (1999).
- 3(3) R. Signorell and F. Merkt, J. Chem. Phys. 110 , 2309 (1999).
- 4(4) H. J. Wörner and F. Merkt, Angew. Chem. 48 , 6404 (2009).
- 5(5) G. Herzberg, Q. Rev. Chem. Soc. 25 , 201 (1971).
- 6(6) R. J. Saykally, and R. C. Woods, Ann. Rev. Phys. Chem. 32 , 403 (1981).
- 7(7) E. Hirota, Chem. Rev 92 , 141 (1992).
- 8(8) E. Hirota, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem. 96 , 95 (2000).