Bonding of Cobalt Atoms, Dimers and Co$_4$ Clusters to Circumcoronene
Tom\'as Alonso-Lanza, \'Angel Ma\~nanes, Andr\'es Ayuela

TL;DR
This study investigates how cobalt atoms, dimers, and clusters interact with circumcoronene, revealing preferred bonding sites, structural arrangements, magnetic effects, and charge transfer characteristics.
Contribution
It provides detailed insights into the bonding configurations, magnetic properties, and charge transfer phenomena of cobalt clusters on circumcoronene, a topic not extensively explored before.
Findings
Cobalt prefers hollow edge sites on circumcoronene.
Cobalt dimers bond perpendicularly to hollow sites.
Co4 clusters adopt a tetrahedral shape with specific bonding arrangements.
Abstract
We study the interaction of circumcoronene molecule CH with cobalt in the form of atoms, dimers and clusters of four atoms. We find that the cobalt atom prefers to be on a hollow site in the edge zone. The cobalt dimer is bonded perpendicularly to another different hollow site in the edge zone. The Co cluster adopts a tetrahedral shape with a face parallel to circumcoronene, placing each of the three atoms over contiguous hollow sites, starting from the edge. Cobalt remains bonded to circumcoronene as Co molecules or Co clusters, rather than spread as isolated atoms, because the cobalt-cobalt interaction is stronger than the cobalt-carbon one. However, the interaction with cobalt clusters induce small magnetic moment on circumcoronene. The magnetic moment of cobalt counterpart is reduced when it is bonded to circumcoronene. There is charge transfer…
Click any figure to enlarge with its caption.
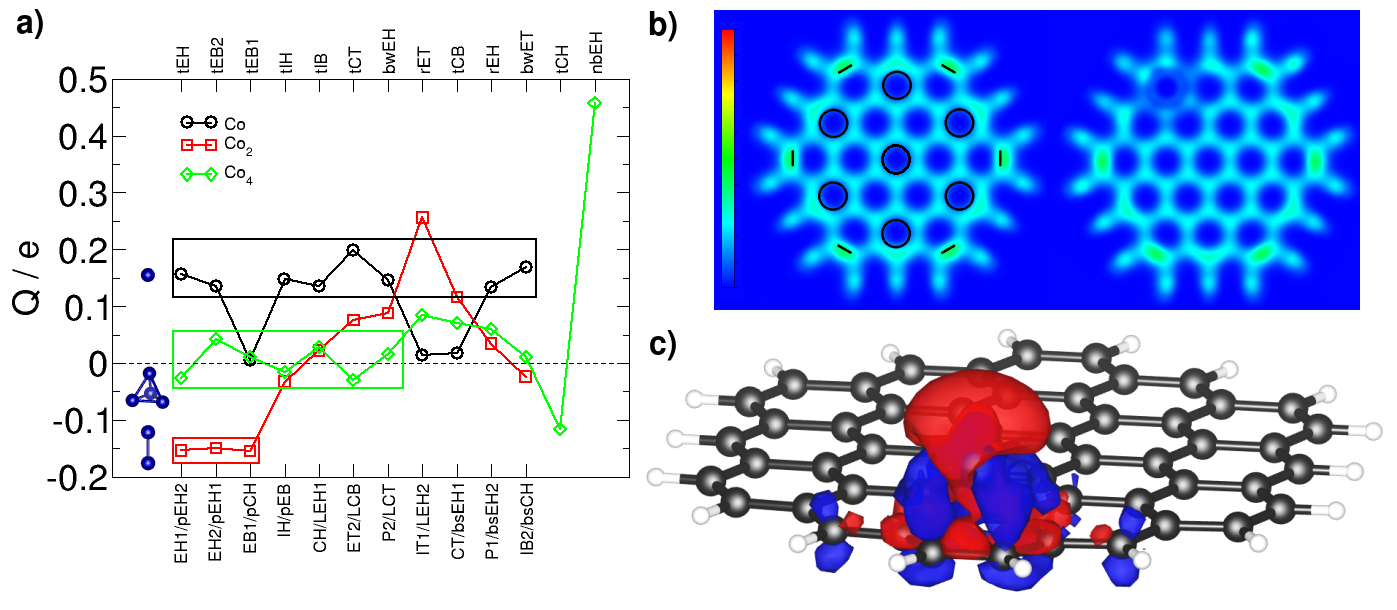 Figure 1
Figure 1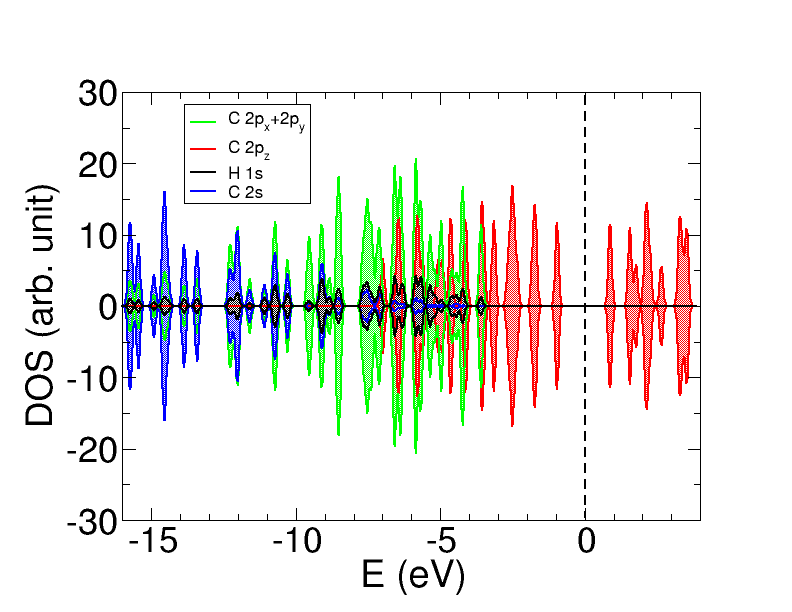 Figure 2
Figure 2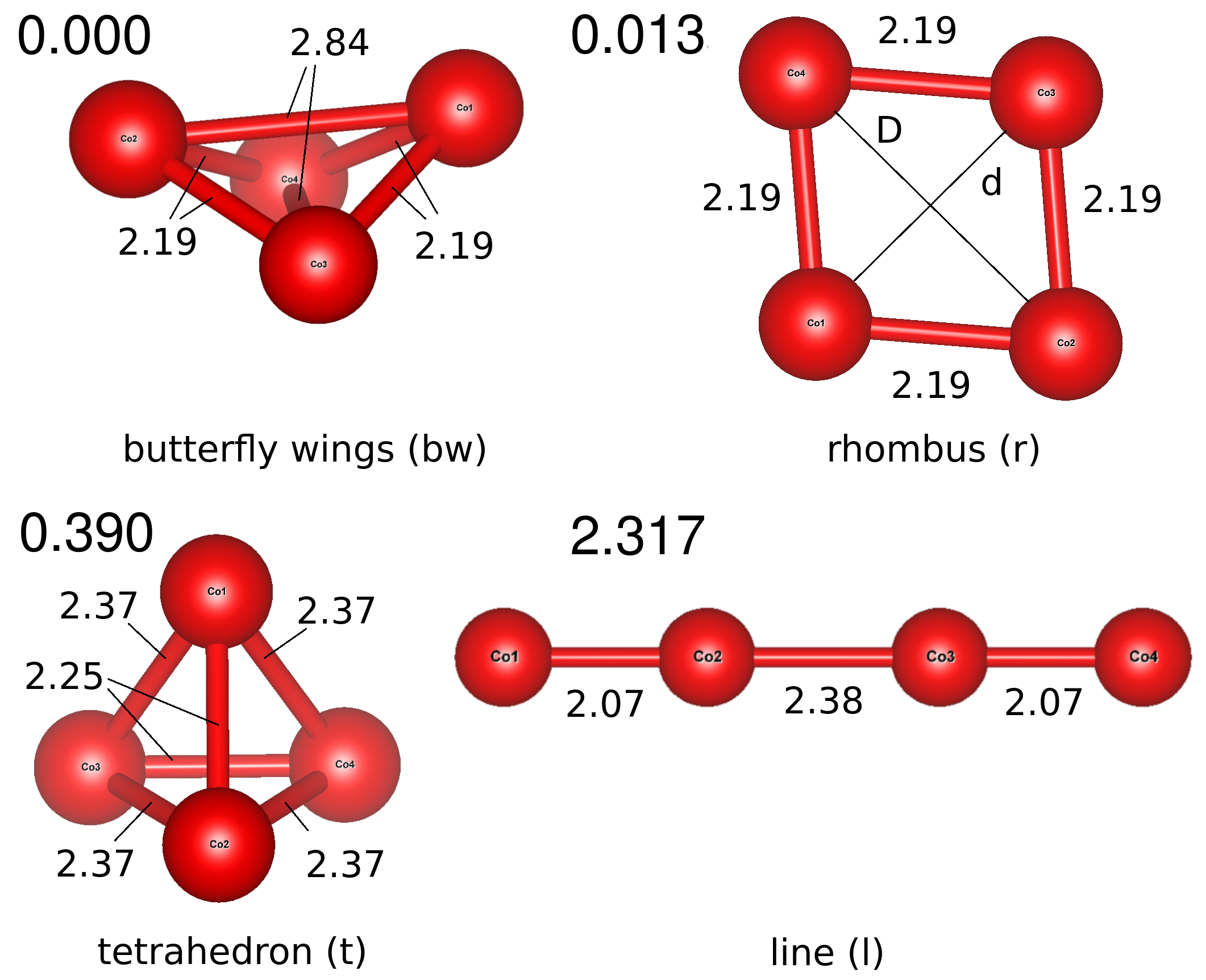 Figure 3
Figure 3 Figure 4
Figure 4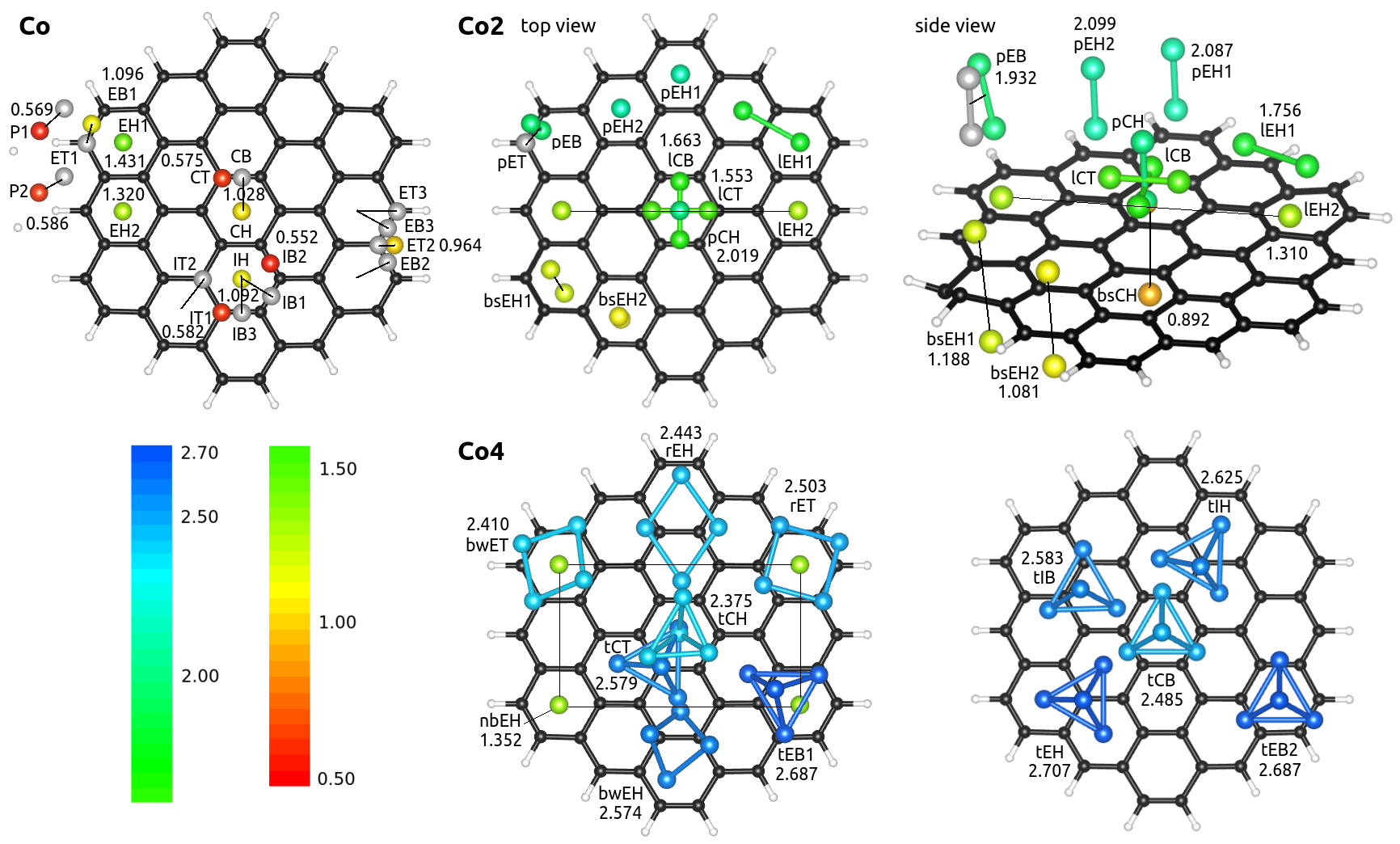 Figure 5
Figure 5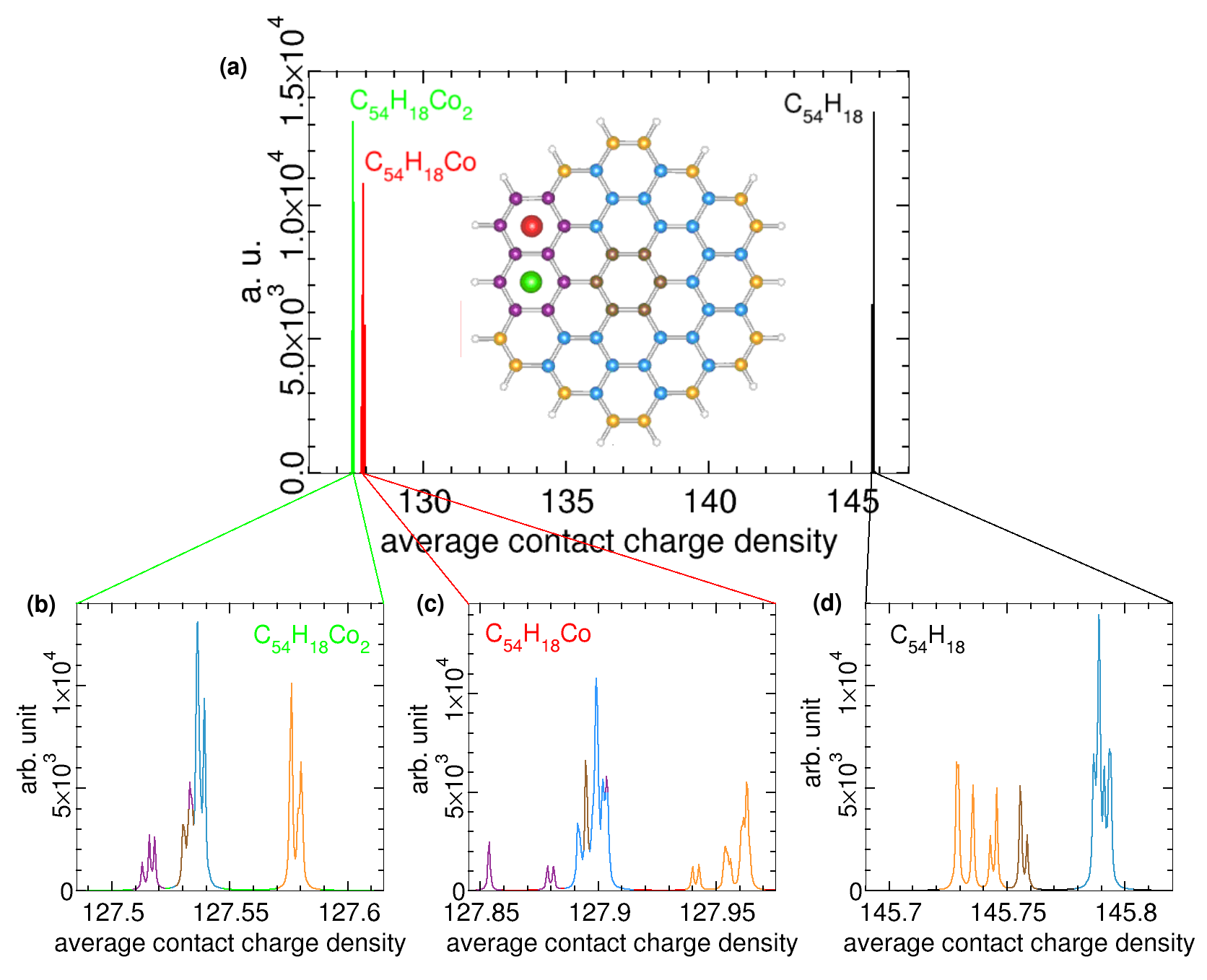 Figure 6
Figure 6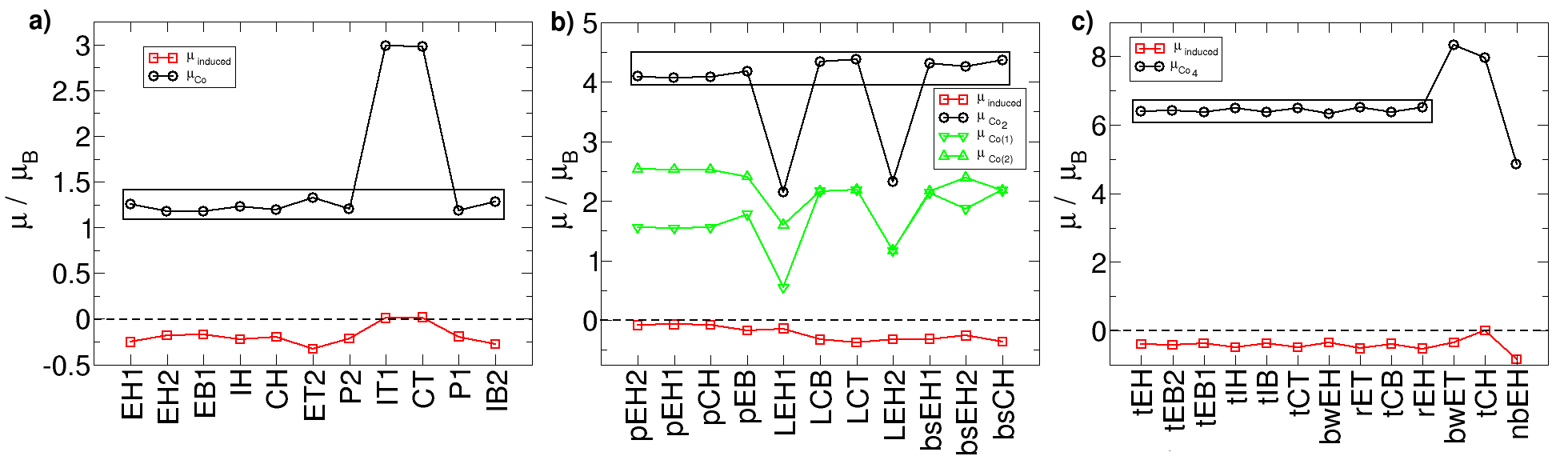 Figure 7
Figure 7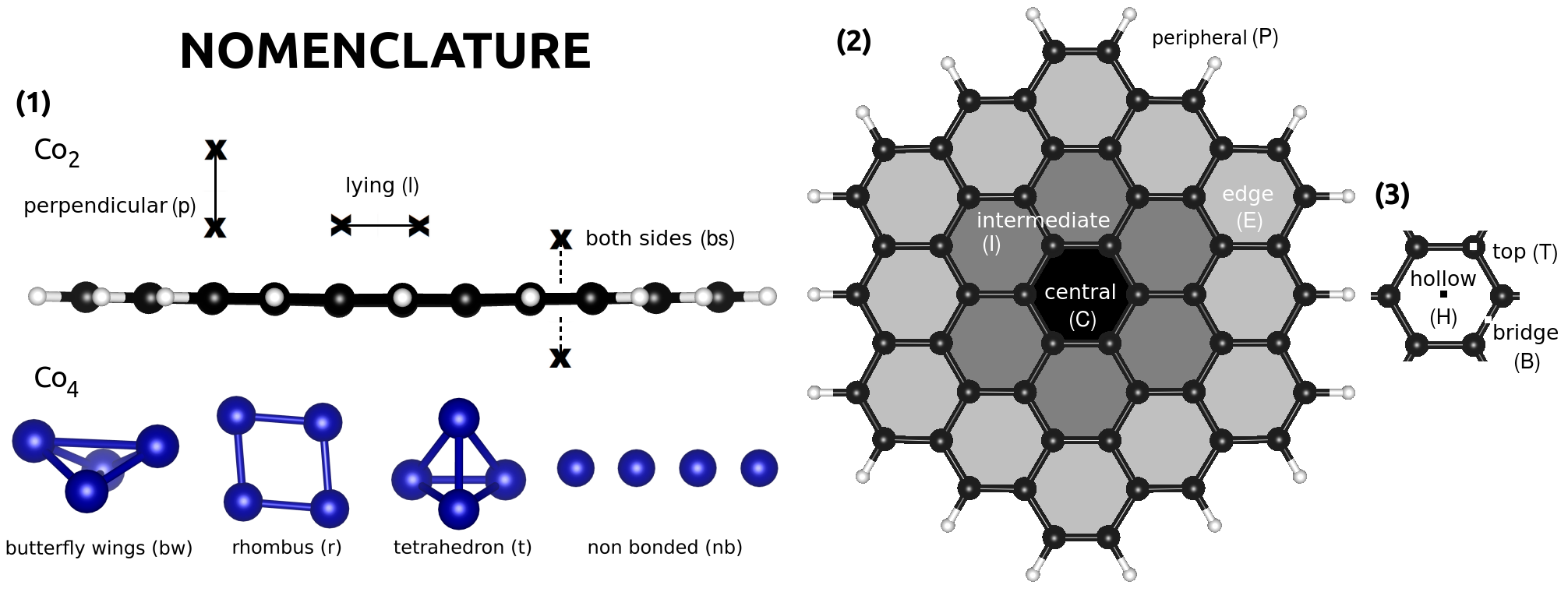 Figure 8
Figure 8| conf | /eV | /Å | /Å | / | / | / | Q/e- | |||||
| CH | 1.000 | 1.516 | 2.138 | 1.000 | 1.151 | -0.151 | 0.11 | |||||
| conf | /eV | /Å | /Å | /Å | /Å | /0 | / | / | / | / | / | Q/e- |
| pCH | 1.779 | 1.794 | 4.161 | 2.281 | 2.368 | 0.00 | 0.000 | -0.078 | -2.007 | 1.929 | 0.078 | -0.216 |
| lCB | 1.700 | 2.289 | 2.289 | 2.141 | 2.168 | 90.00 | 4.000 | 4.308 | 2.154 | 2.154 | -0.308 | 0.083 |
| lCT | 1.598 | 2.291 | 2.291 | 2.137 | 2.191 | 90.00 | 4.000 | 4.328 | 2.164 | 2.164 | -0.328 | 0.062 |
| conf | /eV | /Å / / Co(1)-Co(2)-Co(3)-Co(4) | / | / | / | Q/e- | ||||||
| tCB | 2.547 | 2.112/1.475 - 2.112/1.475 - 2.112/1.475 - 3.989/1.943 | 6.000 | 6.369 | -0.369 | 0.154 | ||||||
| bwCB | 2.442 | 2.254/1.700 - 2.254/1.700 - 3.507/2.403 - 3.507/2.403 | 8.000 | 8.206 | -0.206 | 0.015 | ||||||
| bwCT | 2.425 | 2.258/1.748 - 2.258/1.748 - 3.650/2.384 - 3.650/2.384 | 8.000 | 8.264 | -0.264 | 0.059 | ||||||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHistory and advancements in chemistry
Bonding of Cobalt Atoms, Dimers and Co4 Clusters to Circumcoronene
T. Alonso-Lanza,1, A. Mañanes,2, and A. Ayuela1
1Centro de Física de Materiales CFM-MPC CSIC-UPV/EHU, Donostia International Physics Center (DIPC), Departamento de Física de Materiales, Fac. de Químicas, UPV-EHU, 20018 San Sebastián, Spain
2Departamento de Física Moderna, Universidad de Cantabria, Santander, Spain
Abstract
We study the interaction of circumcoronene molecule C54H18 with cobalt in the form of atoms, dimers and clusters of four atoms. We find that the cobalt atom prefers to be on a hollow site in the edge zone. The cobalt dimer is bonded perpendicularly to another different hollow site in the edge zone. The Co4 cluster adopts a tetrahedral shape with a face parallel to circumcoronene, placing each of the three atoms over contiguous hollow sites, starting from the edge. Cobalt remains bonded to circumcoronene as Co2 molecules or Co4 clusters, rather than spread as isolated atoms, because the cobalt-cobalt interaction is stronger than the cobalt-carbon one. However, the interaction with cobalt clusters induce small magnetic moment on circumcoronene. The magnetic moment of cobalt counterpart is reduced when it is bonded to circumcoronene. There is charge transfer between those systems and its direction depends on the relative position of the cluster with respect to circumcoronene.
I Introduction
Polycyclic-aromatic hydrocarbons (PAHs) such as benzene and coronene (C24H12) are found as by-products in combustion processes Richter and Howard (2000) as well as in the interstellar medium Bohme (1992). PAHs show high stability because of the high delocalization of electrons, which allows these molecules to keep its aromaticity Zhou et al. (2006), key to understand their interesting properties. The properties of larger PAH molecules like circumcoronene C54H18 have also been deeply studied Khanna et al. (2010); Philpott and Kawazoe (2009); Aryanpour et al. (2014); Dias (2002). Circumcoronene have delocalized bonds in the form of seven local sextets and six double bonds, which can be represented in a Clar structure Popov and Boldyrev (2012). Different experimental aspects of circumcoronene have been studied such as interaction with ion beams Forsberg et al. (2013), loss of fragments Bauschlicher Jr and Ricca (2014), nuclear magnetic resonance (NMR) spectra Ikäläinen et al. (2008), and electron currents when applying a perpendicular magnetic field Soncini et al. (2003). As commented above for other PAHs, circumcoronene is specifically thought to be found in the interstellar medium, accounting for the IR spectra that is experimentally obtained. The IR spectra is computed in several studies considering neutral and charged circumronene after adsorbing atoms Bauschlicher and Bakes (2000); Galué and Oomens (2012); Hudgins et al. (2005). Note that atoms can adsorb to circumcoronene on different sites.
PAHs doped with late transition metals are also in the quest for ultimate memories in spintronics Xiao et al. (2009). For smaller PAHs, like benzene and coronene, previous works have studied cobalt adsorption. When a cobalt atom bonds benzene, it is situated over the center of the ring, giving rise to a site BelBruno (2005). It has also been studied how few transition metal atoms interact with several benzene molecules Pandey et al. (2001). It is known to adopt two kind of structures, multiple-decker or bowl-like, depending on the transition element. For late transition elements, such as cobalt, bowl-like structures are preferred Kurikawa et al. (1999). Cobalt atom over benzene is predicted in the hollow site Zhou et al. (2006). The magnetism of the cobalt benzene system is mainly determined by the cobalt atom, which displays 1.1 with a small negative magnetic moment induced in benzene Zhang and Wang (2008). Larger coronene can be produced using laser ablation as in interstellar medium Duncan et al. (1999), characterized using photoelectron spectroscopy Duncan et al. (1999) and collisions with charged ions Ławicki et al. (2011). Furthermore, the interaction between iron and coronene has been studied both theoretically by density functional theory calculations Li et al. (2008) and experimentally by photoelectron spectroscopy Li et al. (2008) and mass selected laser photodissociation Buchanan et al. (1998). Cobalt and coronene interaction has been well studied both theoretically by density functional theory and experimentally using photoelectron spectroscopy Kandalam et al. (2007). For the much larger circumcoronene, iron atoms preferred bonding to hollow sites in the edges Bauschlicher Jr (2009); Simon and Joblin (2010). However, cobalt atoms deposited on circumcoronene remains yet to be investigated, ranging from single cobalt atoms to small cobalt clusters.
Here we study the graphene flake immediately larger than coronene molecule that keeps an hexagonal shape, which is called circumcoronene. We deposit atoms, dimers, and a small cobalt cluster of four atoms over circumcoronene. This hybrid system can display interesting electronic and magnetic properties due to the interaction of the 3d orbitals of the cobalt atoms with the 2p orbitals of the C54H18 molecule. The possible induced magnetic moment in the graphene flake due to the presence of cobalt atoms and additional properties due to the edges could be of importance for the development of graphene-based nanodevices in the field of spintronics.
II Computational details
We present here density-functional-theory calculations that solve the Kohn-Sham equations where the electron density is given as the sum of independent particles. We carry out them mainly with the SIESTA method Soler et al. (2002). We choose the generalized gradient approximation for exchange-correlation with the PBE flavor Perdew et al. (1996) in order to solve these equations self-consistently. The wavefunctions of independent particles are expanded in basis sets of DZP atomic orbitals. We use norm-conserving relativistic Troullier-Martins Troullier and Martins (1991) pseudopotentials for the core electrons of carbon, hydrogen and cobalt atoms. The reference electronic configuration for cobalt has 1.50 electrons in the 4s level and 7.50 electrons in the 3d level. We used fractional charges for the occupations of the pseudopotential attending to the electron configurations for the ground state of the free cobalt atom that we obtained in test ADF calculations. It has been shown that for small transition metal atoms the ground state electronic structure lies in between 3dn-1 4s1 and 3dn-2 4s2 Moroni et al. (1997); Johll et al. (2009). The electron configurations and the value of the radii were thoroughly tested by computing energy differences between all-electron and pseudopotential calculations for several excited states,, such as 3d7 4s2, 3d7 4s1 4p1, 3d7 4s1 and 3d8 4s1. The pseudopotentials were also tested in real calculations, for example, in cobalt bulks. We obtained an hcp structure, with a first neighbors distance of 2.52 Å and a magnetic moment per atom of 1.65 , which are in agreement with experimental results and similar all-electron calculations Myers and Sucksmith (1951). We tested the pseudopotentials in graphene and benzene molecules obtaining reasonable results for their main properties. For our isolated systems we choose a large enough unit cell box of 40x40x40 Å, and atoms are fully relaxed. We have tried different cell sizes for the unit cell to ensure that self-interactions among molecules are negligible. We use the same computational parameters in all the calculations: an electronic temperature of 25 meV and a mesh cutoff of 400 Ry. Additionally, we used another density functional theory codes to validate our results (Amsterdam Density Functional ADF) te Velde et al. (2001), to compute the electron localization function and include the Hubbard term (VASP) Blöchl (1994); Kresse and Furthmüller (1996); Kresse and Joubert (1999) and to compute hyperfine parameters (ELK) Dewhurst et al. (2016). For the VASP calculations we tried values of = 2 and 3 eV, representing a range of reasonable values as found in the literature Piotrowski et al. (2011); Vaugier et al. (2012); Wang et al. (2006); Aykol and Wolverton (2014); Jain et al. (2011) for transition metal systems. We describe in Fig. 2 the nomenclature to specify the different configurations computed for each system with Co atoms, Co dimers, and Co4 clusters.
III Results and discussion
III.1 Stability
III.1.1 Co atom on circumcoronene
We deposit a cobalt atom onto circumcoronene and relax the coordinates starting from several sites. We include top, bridge and hollow sites for the cobalt atom in the three main regions of circumcoronene: central, intermediate and edge. Taking into account that C54H18 has D6h symmetry, the possible sites reduce substantially. The computed configurations are schematically shown in Fig. 3. The binding energies per atom are also included. They are calculated as in eV, where n is the number of cobalt atoms =1,2,4. Note that several of the input geometries suffer large geometry transformations: some cases relax and fall into other proposed geometries, and few relaxed to even new structures. It seems clear from the values of the bonding energy that the hollow sites are in general preferred over the top and bridge sites. For some configurations the cobalt atoms move from the top and bridge sites to the near hollow sites during relaxation, as in the case of CB, IB1, IB3 or IT2 among others. Among the hollow sites in the different zones, we find that the farther from the center of circumcoronene the more stable the hollow site becomes, as given by the sequence CHIHEH2EH1. In the two peripheral configurations, P1 and P2, the cobalt atom bonds laterally to circumcoronene, in the same plane as carbon atoms. The cobalt atoms are in between the hydrogen and carbon atoms, like catalysts. These geometries are the least stable ones.
Cobalt atoms over hollow sites have a height about 1.45 Å over circumcoronene. The Co heights increases up to values close to 2 Å for bridge and top sites, like EB1 and IB2. Anyhow, the cobalt-carbon bond lengths for all the sites are about 2 Å. Note that the P1 and P2 configurations, with the cobalt atom almost replacing an hydrogen atom, have shorter Co-C bond lengths of about 1.8 Å, with the cobalt-hydrogen length about 1.5 Å.
We next compare our results with those of literature. A cobalt atom bonded to benzene on the hollow site is the most stable, having a carbon cobalt distance of 2.02 Å BelBruno (2005). A hollow site is also predicted for a cobalt atom on graphene and graphene nanoribbons Longo et al. (2011) using DFT with the generalized gradient approximation Cao et al. (2010); Johll et al. (2009); Duffy and Blackman (1998); Mao et al. (2008). These calculations show heights over the surface of 1.6 and 1.5 Å, and adsorption energies in the range of 0.97-1.34 and 0.97-2.08 eV, respectively. Quantum Monte Carlo method Virgus et al. (2012) also predicts a hollow site for cobalt atoms in graphene, with a height of 1.49 Å. Using CASSCF to study a cobalt atom over the central hollow site of coronene Rudenko et al. (2012), a height of 1.6 Å is found. A cobalt atom interacting with coronene Kandalam et al. (2007) finds the most stable configuration in a non-central hollow site, like in our most stable EH1 and EH2 configurations, which are non-central hollow sites. The stability and sites of all these previously calculated data are in agreement with our results shown above.
III.1.2 Co2 molecule on circumcoronene
A cobalt dimer bonded perpendicularly to benzene has been proposed as the ultimate magnetic storage device Xiao et al. (2009). In this section we study Co2 dimer on circumcoronene by considering the geometries shown in Fig. 3. Between the two possible types of hollow sites around the edges, the side middle (pEH2) is preferred better over the corners (pEH1). Note that the single cobalt atom prefers to be at the corner site. We find that the cobalt dimer binds to PAH systems perpendicularly at hollow sites close to the edges. The Co heights over the PAH plane for the perpendicular bonds on hollow sites are about 1.6 Å and 3.7 Å. The shortest Co-C distances for each configuration lye within the interval of 2-2.2 Å. The Co-Co distances are about 2-2.1 Å, indicating strong bonds. For the isolated cobalt dimer we obtained approximately 2 Å. Figure 3 shows that disposing the molecule perpendicular is crucial for stability. Although a hollow site is preferred, the pEB bridge configuration is nearly degenerate. The pEB configuration was initially set perpendicular to circumcoronene, but it ends a little tilted, about 13.
Next in stability, we find the configurations lying on circumcoronene. The lEH1 configuration is the most stable among them, and has cobalt atoms over neighbor edge hollows. The distance between cobalt atoms increases up to 2.42 Å, which is approximately 0.3 Å larger than for the perpendicular configurations. The next two most stable configurations place the cobalt molecule parallel to circumcoronene around the central ring, in bridge sites for the lCB case and in top sites for the lCT one, with Co-Co distances of about 2.1 Å. Comparing these two cases with the pCH configuration, which is also on the central ring, we find hollow, bridge and top sites in a decreasing order of stability. The lEH2 case is much less stable with the two cobalt atoms on hollow sites at opposite corners separated by more than 8 Å. The next three configurations spread the two cobalt atoms on equivalent hollow sites at each side of circumcoronene, with large Co-Co distances of 3.2 Å. Anyhow, these configurations together with the lEH2 case are being clearly less stable, which means that the cobalt atoms prefer to bond directly between themselves.
A previous work about coronene Kandalam et al. (2007) found that the most stable configuration have the cobalt dimer perpendicularly on a bridge position at the edge, which could be an analogous to the pEB case, which is indeed highly stable but less than the ground state hollow site. Note that the coronene-cobalt bond length was 2.1 Å Kandalam et al. (2007), which is also similar to 2.06 Å of the pEB case. In agreement with our data, Co2 is perpendicularly bonding graphene over a hollow site with the closest cobalt atom at a height of 1.72 Å and on top a cobalt atom at approximately 2 Å Cao et al. (2010); Johll et al. (2009).
III.1.3 Co4 molecule on circumcoronene
In this section we study the stability of Co4 clusters interacting with circumcoronene. We consider the three most stable geometries of Co4 free clusters, such as butterfly(bw), rhombus(r) and tetrahedron(t), deposited on a corner ring of circumcoronene in the configurations bwET, rET, and tEB1, because the results for Co and Co2 on circumcoronene indicate that on edges they would be more stable. The bw and rhombus structures are commensurate on the top and bridge positions of carbon atoms. The tetrahedron becomes the most stable on circumcoronene.
We searched for the most stable tetrahedron arrangement. The tEH configuration has three cobalt atoms over three different hollows and another cobalt atoms on top of them. It becomes the most stable structure. When the Co4 cluster is adsorbed over the center, circumcoronene remains planar. However, when the Co4 cluster is placed in the edge zone, circumcoronene bend around the cluster. We find some general trends after analyzing all the computed structures. First, moving the cluster from the center to the edges gradually increases the stability. Second, hollow sites are preferred over bridge sites and top sites, which are not even stable in some cases (tCT relax to the hollows). Third, when computing the nbEH geometry we checked that the cobalt atoms prefer to be clustered rather than to spread over circumcoronene separately. All those trends agree with our results for Co and Co2.
The preference for the tetrahedron with one face lying over circumcoronene (tEH) implies large interaction between cobalt and carbon atoms. In the dimer the perpendicular orientation was preferred leaving the farthest cobalt atom almost isolated from carbon atoms. Although it is true that the tetrahedron also isolates one cobalt atom, one can wonder why isolating three cobalt atoms is not preferred (tCH is 0.3 eV higher in energy than tEH). Here there are two trends to be taken into account. First, a cobalt atom over a hollow site is highly stable as seen for EH1. It seems that the more cobalt atoms over hollow sites the larger the stability. Second, commensurating already bonded cobalt atoms over neighbor hollows imply enlargement of Co-Co bond length, which reduces stability. We apply these two ideas to understand Co2 and Co4 cases. For the case of the cobalt dimer, the bond length varies from 2 Å for the free cluster to 2.42 Å for lEH1, which implies an enlargement of 21%. The reduction of stability due to the deformation is not compensated by the possible extra cobalt atom over a hollow site. On the other hand, for the cobalt tetrahedron the bond length varies from approximately 2.3 Å for the free cluster to 2.5 Å for tEH, which implies an enlargement of 9%. As the deformation is lower than for Co2 and, in this case, there are two extra cobalt atoms over hollow sites, the configuration with more cobalt atoms interacting directly with carbon atoms is preferred for the Co4 case, contrary to the Co2 case.
On the other hand, Co4 results show many geometries within an interval of 0.4 eV, quite close in energy. Once the cluster is deposited on circumcoronene, it could slide easily with three cobalt atoms in a plane parallel to circumcoronene. However, additional calculations in order to compute the barriers between those states are beyond the actual scope of this work. Note that the interaction between Co4 clusters and graphene has been studied previously Johll et al. (2011), obtaining a geometry similar to tEH, in agreement with the ground state obtained here.
III.2 Charges and bonding
III.2.1 Charge transfer
We study the charge transfer from Co atoms to circumcoronene which is shown in Fig. 4. In the Pauling scale of electronegativity, carbon and cobalt have values of 2.55 and 1.88, which means that charge transfer from cobalt towards carbon should be expected. Indeed, this happens for most of the cases. Electrons are gained by circumcoronene when a cobalt atom is placed over it, for instance, 0.16 electrons for the ground state (EH1). Similar values are obtained for other configurations except for top sites, such as the IT1 and CT cases with a charge transfer approximately ten times smaller, and for the EB1 configuration with an almost zero value because the cobalt atom is over a double bond. Nevertheless, when Co2 dimer bonds perpendicularly to a circumcoronene hollow (pEH2,pEH1,pCH), about 0.15 electrons pass to cobalt atoms, mainly to the farther atom. When Co2 lies parallel (lEH1,lCB,lCT), circumcoronene slightly gains charge. Note that the lEH2 case presents a large value of 0.256 electrons, as the sum of two EH2 single Co atom configurations. For Co4 the charge transfer is very small in general. Even though, it can be seen that, for tetrahedra, when cobalt atoms are over hollow sites (tEH,tIH,tCT(which relaxed to hollows),tCH) they gain electrons, while when they are over bridge sites (tEB2,tEB1,tIB,tCB) circumcoronene gains electrons. A relatively large value is found for the tCH case with only one cobalt atom bonding a hollow site. As for the lEH2 case, which behaves like two EH1 configurations, the nbEH configuration behaves like four EH1 atom configurations concerning the amount and the sign of the charge transfer. It does not seem to exist a clear correlation between the amount of charge transferred and the stability within each configuration of Co atoms, dimers and clusters.
III.2.2 Bonding
We comment briefly on ideas related the bonding bringing into contact the local charges. We firstly study the preference of cobalt for the circumcoronene edges. We start by considering the aromaticity of the molecule. The well-known Huckel’s rule applies to circumcoronene. Each of the 54 carbon atoms contribute with one electron from the 2pz orbital, so this rule estimates that a planar ring molecule like circumcoronene has aromatic properties when having delocalized electrons with n=13. Following Clar’s rule, the resonance structure of circumcoronene, shown by circles representing aromatic bonds and lines denoting double bonds in Fig. 4b (left part), has the largest number of disjoint aromatic sextets with double bonds found in the edges. However, the electron localization function in Fig. 4b (left part) shows that, although the double bonds are present, there is also charge accumulation at other carbon atoms in the edge, meaning that the Clar structure is not fully reproduced. As a result, circumcoronene has charge accumulation all over the carbon atoms in the edges, explaining the preference of cobalt atoms to be located there. A related argument for the preference for being at the edges would be the loss of symmetry. For the central hollow configuration of the cobalt atom (CH) the 3dxz and the 3dxz levels of cobalt are degenerated, presenting a minority spin level at the Fermi energy. When the cobalt atom is over an edge hollow (the EH1 and EH2 configurations), those levels become different and shift away from Fermi energy to larger energies, and the system gains stability.
Secondly, we comment on the preference for hollow over bridge and top sites. There are two main differences between hollow and top sites: hybridization of cobalt 3d levels with pz states from carbon atoms and filling of cobalt 4s levels. As we show in the next section, the total magnetic moment increases 2 from hollow to top, with some contribution from 4s levels and more than 1 from 3d levels, which suffers a large rehybridization when moving from hollow to top and vice versa. The top site electronic structure is much more similar to that of a free cobalt atom than the hollow one. The large charge reorganization has been argued as the reason for the largest stability for the hollow instead of the top site Johll et al. (2009). Similarly, the preference for a perpendicular bonding of Co2 is explained due to the larger charge change suffered by the farthest cobalt atom; when the dimer is parallel to circumcoronene both cobalt atoms remain similar to free state. The extra energy needed to recover the free state configuration explain the stability of hollow and perpendicular disposition.
We now focus on the charge reorganization for the most stable cobalt atom over circumcoronene (EH1). The ELF in Fig. 4b (right part) shows that charge from the bonds between carbon atoms is removed. This is in good agreement with Fig. 4c, where the red lobes denote weakening of the bonds between the carbon atoms. Furthermore, there is a charge transfer from the upper part of the cobalt atom, denoted by a big red lobe, to the areas which represent the six cobalt carbon bonds, as blue lobes show.
III.3 Magnetism
III.3.1 General remarks
We compute the magnetic moment on each atom as the difference between the Mulliken charge up and the Mulliken charge down. We present in Fig. 5 the magnetic moments of the cobalt atoms and cluster and the induced magnetic moment on circumcoronene. In most of the cases the total magnetic moment is 1 when a cobalt atoms bonds circumcoronene, 4 when it is a cobalt dimer and 6 for the tetramer. We have checked that the net magnetic moment is well preserved when adding the Hubbard term U to Co atoms for the most stable geometry of each case. Among cobalt atoms, those farther to circumcoronene show larger local magnetic moments (2.5 for Co2) because the closer cobalt atoms shield the effect of carbon atoms. For almost all the cases there is negative induced magnetic moment on circumcoronene: -0.25 for EH1, almost zero for the dimer bonding circumcoronene perpendicularly and -0.4 for the tEH1 configuration.
Our results mainly agree with literature. For a cobalt atom over a graphene hollow Cao et al. (2010); Johll et al. (2009); Duffy and Blackman (1998); Mao et al. (2008) the cobalt magnetic moment is 1.15 . Using quantum Monte Carlo Virgus et al. (2012) the total magnetic moment is 1 . It is also 1 when studying a cobalt atom over coronene Rudenko et al. (2012); Kandalam et al. (2007) using CASSCF. The total magnetic moment is reported to be 4 for Co2 over coronene Kandalam et al. (2007) and graphene Cao et al. (2010); Johll et al. (2009), with 1.58 and 1.66 on the close cobalt atom to graphene respectively. For a tetrahedron similar to tEH over graphene Johll et al. (2011) a total magnetic moment of 6 is reported.
Now we comment briefly on the special cases. For a cobalt atom over the IT1 and CT top sites the total magnetic moment is increased to 3 due to the population of the 4s up level. For Co2 it is 2 for lEH1 and lEH2 and for Co4 it is 4 for nbEH. In these three cases the cobalt atoms are largely separated between them, and the value of the magnetic moment can be explained as the sum of two and four individual atoms respectively. For Co4 the bwET and tCH cases display larger magnetic moment (8 ) because they leave more cobalt atoms farther from circumcoronene.
III.3.2 Experimental proposal to measure hyperfine interactions
Electric and magnetic fields internal to the atom interact with the nucleus. This interaction produces as a consequence the hyperfine structure. We compute here hyperfine parameters that could be looked at in experiments. There are two different terms that constitute the hyperfine interaction: quadrupole splitting and magnetic splitting. Apart from those, the spectra is shifted respect to its center of gravity due to the so-called Mössbauer isomeric shift. This shift depends on the chemical environment of the nuclei and it measures the interaction of the s electrons with the nucleus because they have non zero probability of being in the nucleus. The isomer shifts are determined by an average of the charge density within the nuclear volume Shenoy and Wagner (1978). To measure this interaction, we use the ELK code to compute the hyperfine parametersBlügel et al. (1987). Among them we find the average contact charge density valueBlügel et al. (1987), which measures the charge density in a close region to the nucleus. We focus on the value of the average contact charge density expressed in atomic units on each carbon atom for three different cases, namely the ground state structures of C54H18, C54H18Co and C54H18Co2.
In order to give some guidelines that could help to differentiate the presence of cobalt bonded to circumcoronene, we fit a lorentzian to the data obtaining the spectrum for each case. The spectra for the three cases are shown together in Fig. 6 (a), which also contains an inset with a pattern colored correlating the origin of the peaks to their molecule sites. Firstly, let us focus on the spectrum for the bare circumcoronene in Fig. 6(d). It is possible to differentiate between the atoms that are in the edge and the rest of them colored as orange and blue peaks, respectively. There is also a main peak representing the six carbon atoms in the central ring. Within the peaks originated at the edges sites there should be two different peaks which reflects the two different types of atoms. However, they split because the average contact charge density is extremely sensitive to distances and there are tiny differences between some directions. For the blue zone we found four different peaks, which correspond to the four different distances to the center for the blue-colored carbon atoms.
Figure 6(a) shows also the two cases in the presence of Co. The spectra suffers a remarkable shift towards smaller values when cobalt binds circumcoronene. Due to the presence of one more cobalt atom the shift for C54H18Co2 is larger than the one for C54H18Co. We can look in detail for these cases in Fig. 6(b) and (c). To remark that there is an inversion between the two main peaks (orange and blue) when adding cobalt. The blue peak appears on the left part due to larger shift than the orange one. In purple we mark the six carbon atoms that constitute the ring over which the cobalt atom bonded to carbon is located. These peaks are more shifted towards smaller values because of the average contact charge density. Notice that different peaks appear for Co and Co2 due to the different distances to the center.
IV Summary and conclusions
We have studied the interaction of cobalt atoms, dimers and small clusters of four atoms with circumcoronene. Some general trends rise such as the preference of cobalt to bond hollow sites located at the edge of the molecule. Further study revealed the charge accumulation in the edges as the cause of this behavior. We found large charge reorganization that produces reduced magnetic moment, and accounts for the preference for hollow sites. The bonding mechanism over a hollow site consist on weakening carbon carbon bonds with six carbon cobalt bonds. Finally, we propose a possible experiment that could allow to distinguish the presence of cobalt atoms doping circumcoronene based on the isomeric shift attending to the different environment of carbon atoms.
Acknowledgements.
This work has been partially supported by the Projects FIS2013-48286-C02-01-P and FIS2016-76617-P of the Spanish Ministry of Economy and Competitiveness MINECO, the Basque Government under the ELKARTEK project(SUPER), and the University of the Basque Country (Grant No. IT-756-13). TA-L acknowledge the grant of the MPC Material Physics Center - San Sebastián.
Supplemental Information I: SIESTA calculations: Free molecules and clusters
IV.1 Circumcoronene (C54H18)
Among poly-aromatic hydrocarbons (PAH), the circumcoronene molecule is composed of 54 atoms of carbon and 18 atoms of hydrogen, with lateral dimension slightly smaller than 1.5 nm. With respect to the well-known coronene C24H12, the structure of circumcoronene has one more external layer, keeping hexagonal with the carbon dangling bonds saturated by hydrogen atoms in a well-defined D6h symmetry.
Concerning bond lengths in circumcoronene, we found that the position of the C-C bond is related to its length. The bonds around the central point of circumcoronene, taken as reference, have a length of 1.43-1.44 Å, slightly larger than those of pristine graphene (1.42 Å Neto et al. (2009)) and benzene (1.39 Å) Coulson (1947); Pandey et al. (2000). As we move towards the edge, larger and shorter values are alternating. This can be easily understood by the aromatic structure of the molecule. The shortest bond lengths correspond to the double bonds located at the six corners.
Studying the aromaticity of the molecule, the well-known Huckel’s rule applies to circumcoronene. Each of the 54 carbon atoms contribute with one electron from the 2pz orbital, so this rule estimates that a planar ring molecule like circumcoronene has aromatic properties when having delocalized electrons with n=13. Following Clar’s rule, the resonance structure of circumcoronene, shown in Fig. 7, has the largest number of disjoint aromatic sextets with double bonds found in the edges.
About the carbon hydrogen bonds, there are eighteen bonds about 1.11 Å, a value which agrees with the typical bond length for carbon hydrogen bonds in graphane Sofo et al. (2007) and benzene (which is 1.09 Å Pandey et al. (2000)).
To study the stability of the system we compute the bonding energy with respect to the atoms. It is defined as where is the total energy of a single C atom, is the total energy of a single Co atom and is the energy of circumcoronene. We obtain a total bonding energy of 464.982 eV, which is due to 54 carbon atoms and 18 hydrogen atoms, so that per atom it gives 6.45 eV/atom in the order of covalent bonds. This means that the molecule object of study is highly stable. Although the gap is well-known to be underestimated within the exchange- correlation potential used within GGA-DFT, it is about 1.84 eV for the circumcoronene molecule, still far from the zero gap of graphene.
There is no spin polarizations, as seen in Fig 7, because the molecule edges does not have spin. We have computed circumcoronene (both with SIESTA and ADF) setting an initial state with locally polarized states on the edges in a way that has been suggested previously Fernández-Rossier and Palacios (2007), and the final state is locally unpolarized, in agreement with Fernández-Rossier et al Fernández-Rossier and Palacios (2007) predictions. The circumcoronene isolated molecule has zero-net magnetic moment. This agrees with Lieb’s theorem, which for circumcoronene would predict zero spin polarization due to the fact that there is the same number of atoms on each sublattice Fernández-Rossier and Palacios (2007). Nevertheless, it is possible for a system to have partial magnetic moment on their atoms and to have a value of zero for the total magnetic moment of the system. Experimentally it is possible to obtain the spatial distribution of the edge states spins in the graphene nanosheets using the electron spin resonance (ESR) technique at a temperature lower than 20 K Joly et al. (2010). The shape of the nanosheet determine both the local magnetic moments and the net magnetic moment. In this type of molecules, the magnetic moment would be localized in the edges, and it would be due to spin-polarized non-bonding states. When the edges are magnetic, there is a strong ferromagnetic (FM) coupling between the edge states that belong to the same sublattice and a weaker FM or antiferromagnetic (AFM) coupling between those of different sublattice. The atoms in a zigzag edge belong to the same sublattice and so they have ferromagnetic coupling. If we apply it to circumcoronene, all the atoms of each side in a hexagonal shape belong to the same sublattice, and the atoms in the neighbor side to another sublattice. Thus, if we found local magnetism in the edge of circumcoronene we could expect to have each side alternatively polarized. However, the existence of local magnetic moments depends not only on the size of the nanosheet but also in the shape. Kumazaki et al Kumazaki and S. Hirashima (2008) used a Hubbard model to study magnetism on the edges, obtaining that vacancy states in the same sublattice can generate local magnetic moments; they proposed that three consecutive vacancies of the same sublattice are enough to obtain local magnetic moment. Attending to this, circumcoronene should present local magnetic moment. However, Fernández-Rossier et al Fernández-Rossier and Palacios (2007) proposed a minimum size of eight atoms per side to find locally polarized states Fernández-Rossier and Palacios (2007). Since circumcoronene has three edge atoms along one edge, it is expected to find still locally unpolarized states.
We analyze Fig 7 in detail looking at the projected density-of-states for the s states for the carbon and hydrogen atoms and for the p states for the carbon atoms. The s states from hydrogen and carbon are found at the same low energies because they are hybridized. We differentiate between the 2p states that are in the plane (2px and 2py) and the 2p states that are perpendicular to circumcoronene (2pz). The p-type states lying on the plane of circumcoronene are also hybridized with the s-states, in the so called sp2 hybridization, leaving the 2pz non-hybridized. The 2pz, located at higher energies, are both occupied and empty states around the Fermi level, so they would be held responsible for bonding cobalt atoms.
IV.2 Free cobalt clusters
Co atom. We compute the cobalt atom using the SIESTA code and we obtain a magnetic moment of 3 . We find that the 4s and 5d levels have lower energy when they are occupied by 1.6 and 7.4 electrons, respectively. We use this configuration to generate Co pseudopotential.
*Co2 molecule. * Using the generated cobalt pseudopotential, we compute the cobalt dimer. We obtain a relaxed bond length of 2.036 Å, a bonding energy of 1.139 eV, and a magnetic moment about 5 . Johll et al Johll et al. (2009) report a bond length of 1.94 Å and a total magnetic moment of 4 for the cobalt dimer performing DFT calculations using ultrasoft pseudopotentials. Seifert et al Xiao et al. (2009) obtain also a value of 4 for the magnetic moment of the cobalt dimer, using an all-electron full-potential local-orbital scheme (FLPO) code. But the magnetic moment of the cobalt dimer has also been reported to be 5 when performing DFT calculations using the BLYP functional Zhang and Wang (2008). See next Appendix searching on the comparison with our ADF test results.
Co4 cluster
We check four different geometries for the free Co4 cluster in order to find the low lying isomers with minimum energy: two different tetrahedra, a square and a line, shown in Fig. 8. The regular tetrahedra has initial edges 2.25 Å long, following the equilibrium geometry in Ref. Yoshida et al. (1995), where a distorted tetrahedron had three edges of 2.25 Å and the other three with 2.37 Å. The input regular square has sides of 2.1 Å, and in the linear structure all the cobalt atoms are separated by 2.1 Å. The most stable structure is a non-regular tetrahedron that we called butterfly wings (BW) because of its shape. Note that it is obtained by fully relaxing the input regular tetrahedron, which suffers a very large structural transformation. It has a bonding energy per atom of 2.078 eV. The second most stable isomer is the square with a close bonding energy of 2.075 eV, nearly degenerated, which ends in a rhombus like structure. The third most stable isomer is a less distorted tetrahedron with a bonding energy of 1.980 eV. All these isomers are largely preferred over the linear structure, which presents quite lower bonding energy, 1.499 eV. Since the linear structure is much less stable, it means that maximizing the number cobalt-cobalt bonds increases stability. However, in the most stable structure after relaxation there are two kinds of bonds: ones that becomes much larger and other much shorter Datta et al. (2007), the latter allow for large 3d bonding interaction. This effect looks like Jahn-Teller distortion.
The total magnetic for tetrahedra isomers is about 10 except for the less distorted tetrahedra, which has a slightly larger value of almost 11 . The four atoms are equivalent from the magnetic point of view, except the BW isomer with local moment values of 2.500 and 2.672 . For the rhombus structure, the atoms opposed by the large diagonal have a magnetic moment of 2.43 , while the atoms opposed by the shorter diagonal have close values of 2.58 For the linear structure, the atoms at the ends have a larger value of the magnetic moment (2.626 ) than the atoms in the center (2.374 ).
All these findings are in agreement with a large amount of publications on transition metal clusters and, specifically, on cobalt clusters. Focusing on Co4 cluster, the most stable energy structure is a tetrahedron Yoshida et al. (1995); Datta et al. (2007); Rodríguez-López et al. (2003); Johll et al. (2011); Andriotis and Menon (1998) as we obtain in our calculations. However, this tetrahedron structure is reported to have differences. For example, in Ref. Rodríguez-López et al. (2003) the tetrahedron cluster remains non-distorted with a magnetic moment per atom of 2.99 , and an average bond distance of 2.34 Å. Andriotis et al Andriotis and Menon (1998) obtained a tetrahedron with bond lengths between 2.61-2-80 and a magnetic moment of 2.50 on each cobalt atom. In Ref. Datta et al. (2007) they also obtained that the most stable is a highly distorted tetrahedron followed by a rhombus, the two structures being much more stable than the linear one. Concerning the magnetic moment Datta et al. (2007), all of the cobalt atoms in the tetrahedron and the rhombus have the same value, 2.5 . Johll et al Johll et al. (2011) proposed a structure with two bond lengths of 2.14 and 2.74 Å, in agreement with the one we found here, and also the total magnetic moment (10 ).
Supplemental Information II: Full electron ADF results brought into contact with SIESTA ones
In this section we begin by showing test results obtained using the ADF code. We used a frozen core treatment of the inner shell. The equations are solved using localized basis set of Slater type orbitals for the wave functions and the generalized gradient approximation for the exchange and correlation energy, using the revPBE expression. For circumcoronene with Co atoms, Co dimers, and Co4 clusters we test some of the configurations with cobalt around the central hollow. These geometries are the most appropriate to compare results with those of graphene because the central hollow is the farthest position from the edges, and in some way it can be expected a compensation of the border effects due to the symmetry. Obtained with a code that uses all electron basis sets, these calculations serve us next to check the results from the SIESTA code calculations in order to assess the validity of the pseudopotentials employed. We collect the ADF results for these center configurations in Table 1.
We start reviewing the magnetic properties focusing first on the free systems. For the circumcoronene molecule we obtained a non-magnetic equilibrium state, with no localized magnetic moment on edge atoms, which agrees with results of previous work in base to the size of circumcoronene Fernández-Rossier and Palacios (2007). Note that it is an aromatic molecule following Clar’s rule Popov and Boldyrev (2012). For the free cobalt atom the magnetic moment in the equilibrium state is 3 . When we allowed charge division, we obtain that an electron configuration like stabilizes the system. Based on these results we generate the pseudopotential that we are using here in SIESTA calculations. Concerning the cobalt dimer we obtained a bond length of approximately 2 Å, with a total magnetic moment of 4 , as in Ref Xiao et al. (2009). However, SIESTA calculations obtain values about than 5 . The bonding energy EB for the cobalt molecule is 1.757 eV. For the Co4 cluster, the magnetic moment is 10 . The structure is called butterfly wings (bw) after its shape. Almost square in shape, the bond lengths are 2.208 Å and 2.859 Å. SIESTA drops values quite similar, of 2.19 Å and 2.84 Å. The values of the magnetic moments on each atom are the same at geometrically equivalent sites because there is no charge transfer found between cobalt atoms. The bonding energy EB for the four cobalt atoms is 2.440 eV.
In order to understand why we do not get the same result as with ADF, we have checked a large amount of radii for the pseudopotential generation without getting closer to the 4 value for Co2 molecule. Even when we reduced the radii as much as possible to have a hard but highly transferable pseudopotential, we obtain the desired value of magnetization, but we spoiled the good results for larger systems, as Co4 clusters or cobalt bulk. We check that choosing the pseudopotential relativistic or not is important in this particular case. We obtain that for non-relativistic pseudopotentials the order of the levels corresponds to a 4 state, changing the magnetization because a very small broadening would be required to get closer to the 4 value. Then, we create a pseudopotential with 3p electrons included as valence electrons, but we did not yet get the desired 4 value. Finally, by including the 3s electrons we got the reported as correct magnetic moment value Xiao et al. (2009). To get this value of the magnetic moment the FPLO code requires to include the 3s electrons as valence electrons. However, we note that another option to get closer to the 4 value is to change the electric configuration from 4s1.53d7.5 to 4s13d8. In this way we obtain values slightly larger than 4 , which also improves previous results. From all this discussion, the conclusion is that the cobalt dimer stands forward as critical to be modeled using pseudopotentials. To obtain the magnetic moment of 4 requires reducing the number of core electrons, which in fact is moving towards a full-electron calculation, as it is the case of using ADF or FLPO codes Xiao et al. (2009). Anyhow, for larger systems, pseudopotentials are not so crucial, and similar results are obtained with different optimized pseudopotentials. In this work we are using the pseudopotential we obtained that provided the smallest energy differences among several electronic configurations during the tests we carried out. Including 3s and 3p electrons would improve our results for larger systems, but only slightly; this improvement does not justify the loss of computation speed, taking into account that we chose SIESTA on purpose to study in detail many adsorption geometries and to discuss general trends, more than focusing on a specific configuration. Nevertheless, this issue with Co2 molecule shows useful to know the limitations of our choosen pseudopotential.
We next proceed to deposit Co atoms, Co molecule and Co4 clusters around the center of circumcoronene. The bonding energy is computed as , with n=1,2,4. In the central ring of circumcoronene, Co atom is at the central hollow site (CH) with a height of 1.5 Å, establishing six bonds to the carbon atoms with a bond length of 2.13 Å. The magnetic moment of the system amounts to 1 . The SIESTA results for this configuration present similar values for bond heights and lengths of 2.105 Å and 1.454 Å respectively.
For Co2 on circumcoronene we tried three configurations in top, bridge, and hollow sites (lCT, lCB and pCH). The most stable is the perpendicular pCH hollow configuration, with a cobalt atom placed at the centre of the ring, with a bond length of 2.28 Å, slightly larger than in SIESTA results where it was 2.201 Å. The height over circumcoronene is 1.77 Å, comparable to 1.668 Å obtained with SIESTA. However, the cobalt-cobalt length becomes 2.37 Å obtained with ADF shorter from 2.12 Å with SIESTA. These trends propagate the magnetic moment of Co molecule once adsorbed on circumcoronene. For the ADF calculation, the magnetic moment is zero because the cobalt atoms are anti-ferromagnetically coupled, while it becomes ferromagnetic with a value of 4 in the SIESTA calculation. Table 1 gives the adsorption energy of the pCH configuration in ADF code about 0.042 eV, which indicates that the bonding process could be considered to be physisorption more than chemisorption, as in the rest of cases. Stretching slightly the distance Co-Co in SIESTA calculations we end in the same zero antiferromagnetic values; this indicate that in the search in previous ADF calculations of Co molecules on circumcoronene, we have overlooked the minimum energy for the ferromagnetic configuration.
We obtained that placing the cobalt dimer perpendicularly over the central hollow site (pCH) results in a more stable geometry than placing it lying on the bridge (lCB) or top (lCT) sites around the central hollow site. In fact the adsorption energies for the lCB and lCT configurations are negative, which indicates that not only the pCH configuration is more stable but that is preferred over the free atoms. The same stability order is reproduced with the SIESTA calculations in Table III for those three configurations.
We last compare ADF geometries with those obtained using SIESTA. In the case of lCB configuration, the cobalt height over circumcoronene differs by about 0.1 Å, thus the cobalt-carbon bond length is slightly larger. Anyhow, the values of the magnetic moment are quite similar within 0.1 . For the lCT configuration, the cobalt height and the cobalt-carbon bond length are shorter, while the cobalt-cobalt bond length is slightly larger. The magnetic moment values are also similar, but in ADF results the magnetic moment of cobalt atoms and the induced magnetic moment on circumcoronene are smaller. For both lCT and lCB configurations, the total magnetic moment in ADF calculations is 4 , which is the same to 4.0 obtained in SIESTA calculations.
We compute Co4 clusters in the circumcoronene center. We tried three configurations starting from the found ground isomer of Co4 clusters. Placing the Co cluster around the central hollow site, the bw structure lies (i) with an edge parallel to circumcoronene and its two cobalt body atoms over bridge sites in the bwCB case or (ii) on top sites in the bwCT one, or (iii) joined by a wing face with the three cobalt atoms on alternate bridges. The third case becomes the most stable after structure relaxations. The outer cobalt atom moved towards the side center and the hollow site to build a regular-like tetrahedron. This outer atom ends with a cobalt-carbon distance of 2.11 Å, similar to the one obtained in SIESTA (2.05 Å), and the magnetic moment is 6 , identical in both codes.
For the bwCB and bwCT configurations the values of the magnetic moment in ADF and SIESTA are also similar, but now two pairs of cobalt atoms behave symmetrically in the same way. In all the cases the charge transfer goes from cobalt atoms to carbon atoms. The bwCB and bwCT configurations have a total magnetic moment of 8 , larger than for atoms in the CB configuration. The adsorption energy for the bwCB case is almost zero and negative for bwCT. This means that the cobalt Co4 cluster prefers to be in the free-state rather than to be adsorbed in those unstable configurations.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Richter and Howard (2000) H. Richter and J. Howard, Prog. Energ. Combust. 26 , 565 (2000).
- 2Bohme (1992) D. K. Bohme, Chem. Rev. 92 , 1487 (1992).
- 3Zhou et al. (2006) J. Zhou, W.-N. Wang, and K.-N. Fan, Chem. Phys. Lett. 424 , 247 (2006).
- 4Khanna et al. (2010) R. Khanna, A. Waters, and V. Sahajwalla, Comput. Mater. Sci. 49 , S 108 (2010).
- 5Philpott and Kawazoe (2009) M. R. Philpott and Y. Kawazoe, J. Chem. Phys. 131 , 214706 (2009).
- 6Aryanpour et al. (2014) K. Aryanpour, A. Shukla, and S. Mazumdar, J. Chem. Phys. 140 , 104301 (2014).
- 7Dias (2002) J. R. Dias, J. Mol. Struct. THEOCHEM 581 , 59 (2002).
- 8Popov and Boldyrev (2012) I. A. Popov and A. I. Boldyrev, Eur. J. Org. Chem. 2012 , 3485 (2012).