Probing molecular potentials with an optical centrifuge
A.A. Milner, A. Korobenko, J.W. Hepburn, V. Milner

TL;DR
This study employs an optical centrifuge to excite molecules to extremely high rotational states, enabling precise probing of their potential energy surfaces at large inter-nuclear distances through time-resolved spectroscopy.
Contribution
It demonstrates the use of an optical centrifuge for ultra-high rotational excitation and compares experimental revival periods with theoretical potential models.
Findings
Successful excitation of molecules to high rotational quantum numbers.
Detection of rotational revivals indicating bond stretching.
Agreement between measured revival periods and Morse-cosine potential calculations.
Abstract
We use an optical centrifuge to excite coherent rotational wave packets in NO, CS and OCS molecules with rotational quantum numbers reaching up to J=465, 690 and 1186, respectively. Time-resolved rotational spectroscopy at such ultra-high levels of rotational excitation can be used as a sensitive tool to probe the molecular potential energy surface at inter-nuclear distances far from their equilibrium values. Significant bond stretching in the centrifuged molecules results in the growing period of the rotational revivals, which are experimentally detected using coherent Raman scattering. We measure the revival period as a function of the centrifuge-induced rotational frequency and compare it with the numerical calculations based on the known Morse-cosine potentials.
Click any figure to enlarge with its caption.
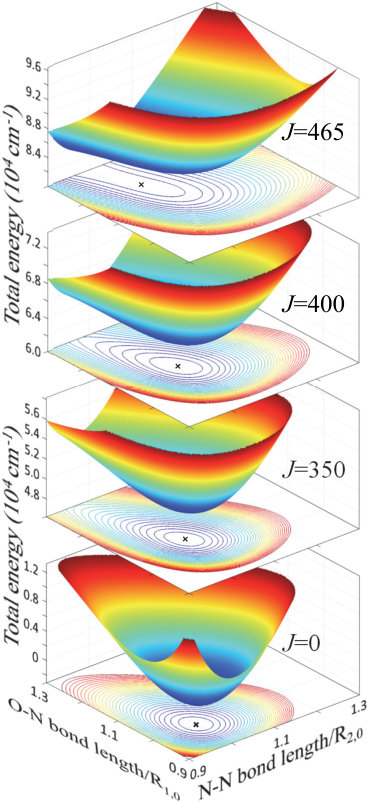 Figure 1
Figure 1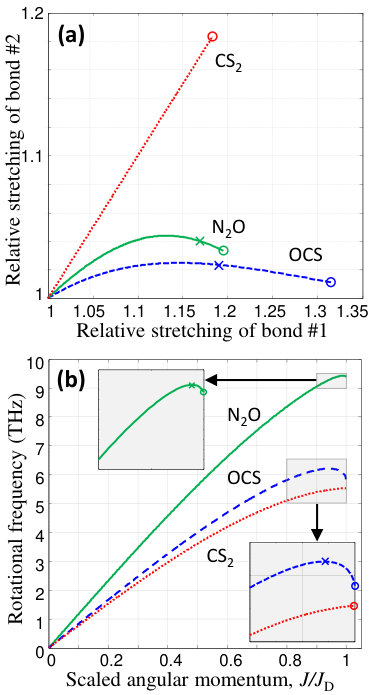 Figure 2
Figure 2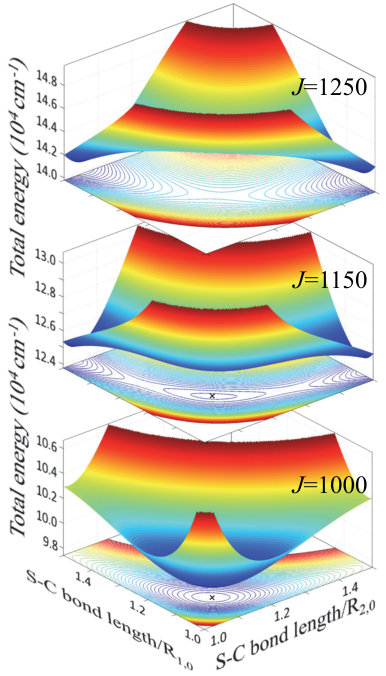 Figure 3
Figure 3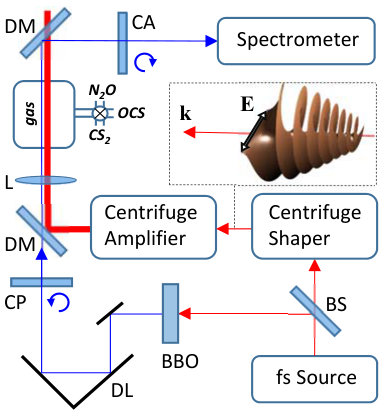 Figure 4
Figure 4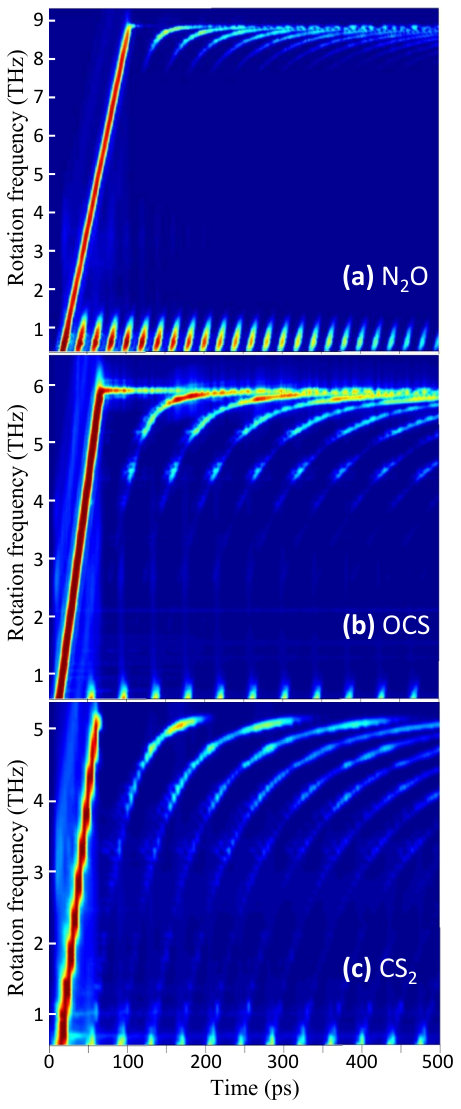 Figure 5
Figure 5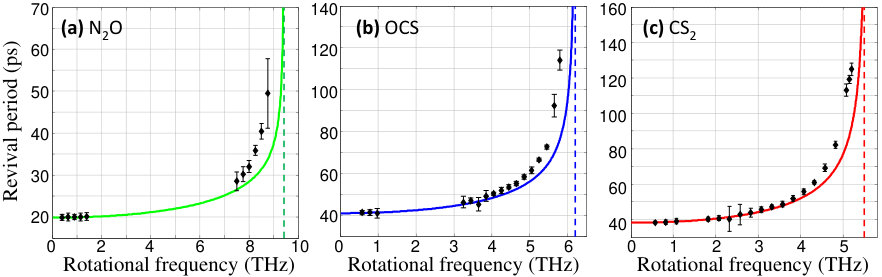 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Probing molecular potentials with an optical centrifuge
A. A. Milner
A. Korobenko
Department of Physics & Astronomy, The University of British Columbia, Vancouver, Canada
J. W. Hepburn
Department of Chemistry, The University of British Columbia, Vancouver, Canada
V. Milner
Department of Physics & Astronomy, The University of British Columbia, Vancouver, Canada
Abstract
We use an optical centrifuge to excite coherent rotational wave packets in , and molecules with rotational quantum numbers reaching up to , 690 and 1186, respectively. Time-resolved rotational spectroscopy at such ultra-high levels of rotational excitation can be used as a sensitive tool to probe the molecular potential energy surface at internuclear distances far from their equilibrium values. Significant bond stretching in the centrifuged molecules results in the growing period of the rotational revivals, which are experimentally detected using coherent Raman scattering. We measure the revival period as a function of the centrifuge-induced rotational frequency and compare it with the numerical calculations based on the known Morse-cosine potentials.
Potential energy surfaces (PES) are central to almost every aspect of molecular dynamics as they govern both the motion of atoms inside a single molecule Green, Moore, and Polik (1992) as well as collisions and chemical reactions between several molecular reagents Polanyi and Zewail (1995). Mapping out PES of polyatomic molecules, possessing many degrees of freedom, is a difficult task which becomes especially challenging far from the equilibrium, where multiple vibrational and rotational modes become strongly coupled. A common approach consists of expressing the ro-vibrational energy of a molecule as a function of internal molecular coordinates, calculate the frequencies of vibrational transitions and adjust the expansion coefficients so as to match the calculated frequencies with experimental observations. The higher the energies of the observed ro-vibrational transition, the wider the region of the potential energy surface which can be probed and accurately mapped out, the richer the molecular dynamics available for quantitative studies.
A great deal of work has been done on triatomic molecules, for which precise spectroscopic information about highly excited vibrational transitions is readily available. For the molecules studied here – , and , vibrational energies up to approximately cm*-1*, cm*-1* and cm*-1*, respectively, have been reported. In a series of works, Zúñiga et al. Zúñiga et al. (1999, 2000, 2002) developed optimal generalized internal coordinates to fit the ro-vibrational spectrum of these molecules by approximating their potential energy surface with a fourth-order Morse-cosine expansion Jensen (1988):
[TABLE]
where and are the lengths of the two molecular bonds, is the angle between them, are the expansion coefficients and
[TABLE]
with the subscript ‘0’ denoting the equilibrium value for the corresponding coordinate in the ground ro-vibrational state. Constants and determine the stiffness of the respective bonds.
In this work, we use high-energy rotational, rather than vibrational, excitation to study the molecular potentials at the limit when the molecular bonds are stretched far beyond their ground-state values. We employ the technique of an optical centrifuge Karczmarek et al. (1999); Villeneuve et al. (2000) to spin the molecules of interest to ultra-high states of angular momentum . The total energy of a rotating molecule can be expressed as
[TABLE]
where is the molecular moment of inertia and is the reduced Planck’s constant. As the angular momentum increases, the second term in the above expression pushes up the equilibrium bond lengths and , corresponding to the minimum of the total energy, to higher and higher values, eventually resulting in dissociation. The effect is illustrated in Figure 1, where we plot the potential energy surfaces of at different levels of rotational excitation, calculated with Eq.3 and the Morse-cosine PES coefficients from Ref. 3. While not much distortion is happening below , the shape of the PES changes rapidly at higher values of the molecular angular momentum, until the potential minimum disappears at the dissociation limit . Notably, before the molecule dissociates, its total energy increases to about cm*-1*, almost an order of magnitude above the level typically accessible through vibrational excitation Campargue et al. (1995).
Rotational dissociation by an optical centrifuge has been demonstrated experimentally with a diatomic chlorine Villeneuve et al. (2000), and theoretically studied for both the diatomic (Cl2, Ref.7; 10) and triatomic (HCN, Ref. 11) molecules. In both cases, it was predicted that the majority of molecules undergoing forced accelerated rotation will be ejected from the centrifuge prior to reaching the dissociation energy due to the Coriolis force. The latter causes the molecular axis to turn away from the laser field polarization and eventually fall behind the rotating trap. Here, we analyze the molecular dynamics in the vicinity of this Coriolis wall both numerically and experimentally, in several centrifuged triatomic molecules.
Figure 2(a) shows the calculated stretching of both molecular bonds in , and with the rotational quantum number increasing from to the calculated dissociation limit and 1186, respectively. The corresponding dissociation energies are cm*-1*, cm*-1* and cm*-1*. As anticipated for a linear molecule, the equilibrium angle between the bonds remains zero regardless of . The main goal of this work is to demonstrate how such bond stretching under ultrafast centrifuge spinning can be used for probing high-energy regions of the potential energy surfaces. Although neither internuclear distances nor angular momenta are measured directly in our experiments, we accomplish the task by investigating the periodic dynamics of the centrifuged molecules as a function of the rotational frequency, controlled by the centrifuge. Our analysis is carried out as follows.
Given the rotational energy spectrum , the classical rotational frequency at any can be calculated as
[TABLE]
The results of these calculations in the simplest approximation which neglects the zero-point vibrational energy, i.e. , are shown in Fig.2(b). The growth of with the molecular angular momentum is sub-linear, indicating strong centrifugal distortion. Similarly to the previously discussed Karczmarek et al. (1999); Hasbani et al. (2002) behavior of Cl2 and HCN, the rotational frequencies of and fall off above the critical values of (green solid) and (dashed blue), respectively. The corresponding frequency maxima of THz and 6.2 THz, marked with crosses in Fig.2(b), represent the Coriolis wall on the way of accelerated molecular rotation. Beyond this wall, the distance between consecutive rotational levels starts decreasing and the molecules can no longer adiabatically follow the centrifuge field.
Interestingly, the rotational frequency of increases monotonically (dashed red line), indicating that the molecule can climb the rotational ladder all the way to the dissociation frequency THz without escaping the centrifuge. This conclusion, supported by our experimental observations (discussed below), can be interpreted as follows. In asymmetric linear triatomic rotors, the centrifugal force stretches predominantly one of the two bonds (O–N, S–C and N–C in , and HCN, respectively), whereas the other one extends to a much lesser degree and even starts shrinking with the increasing rotational frequency [see Fig.2(a)]. The quickly stretching bond is responsible for the strong Coriolis force, which makes the molecule lag behind, and eventually fall off, the centrifuge without dissociating.
The symmetry of , on the other hand, effectively stiffens the molecule with respect to the centrifugal pull. The lengths of both S–C bonds increase slower than in the asymmetric case, which results in the weaker Coriolis force and better stability in the centrifuge. The difference between the symmetric and asymmetric rotors can be better appreciated by comparing the respective potential energy surfaces near dissociation. While the Gaussian curvature (the product of two principal curvatures) of the potential well of remains positive with increasing angular momentum (Fig.1), its sign changes abruptly when the PES of develops saddle geometry at (Fig.3). The subsequent sudden elongation of one of the S–C bonds, even if accompanied by strong Coriolis force, leads to an inevitable dissociation of the molecule. Although the above classical picture does not account for the quantum tunneling out of the well, the latter will only expedite the dissociation process rather than prevent it.
Since the critical frequency values ( and ) are within the reach of our optical centrifuge ( THz), we were able to study the described behavior experimentally. After the molecules are spun up and released from the centrifuge, we monitor their dynamics by means of coherent Raman scattering Korobenko, Milner, and Milner (2014). An excited rotational wave packet undergoes periodic revivals, separated in time by Leichtle, Averbukh, and Schleich (1996); Seideman (1999)
[TABLE]
Fractional revivals may also occur (as is the case here), depending on the particular structure of the wave packet Averbukh and Perelman (1989). By comparing the experimentally found dependence of the revival time on the rotational frequency, , with the one calculated using the theoretical energy spectrum , the validity of the latter can be readily assessed.
The experimental setup, shown in Fig. 4, is similar to that used in our previous work Korobenko, Milner, and Milner (2014). A beam of femtosecond pulses from a regenerative Ti:Sapphire amplifier (800 nm, 1 KHz repetition rate, 30 nm full width at half maximum) is split in two parts. One part is sent to a “centrifuge shaper”, implemented according to the original recipe of Karczmarek et al. Karczmarek et al. (1999), which converts the input laser field into the field of an optical centrifuge, schematically illustrated in the inset. The shaper is followed by a home built Ti:Sapphire multi-pass amplifier (10 Hz repetition rate), which boosts the energy of centrifuge pulses up to 30 mJ/pulse. The pulses are about 100 ps long and their linear polarization undergoes an accelerated rotation, reaching the angular frequency of 10 THz by the end of the pulse. The second (probe) beam is frequency doubled in a nonlinear BaB2O4 (BBO) crystal, time delayed by means of a controllable translation stage and combined with the centrifuge beam on a dichroic mirror. Both beams are focused into a cell, filled with either , or gas, with a m lens.
We detect the dynamics of the centrifuged molecules using time-resolved coherent Raman spectroscopy. The centrifuge-induced coherence between the rotational states and (where is the projection of on the propagation direction of the centrifuge field) results in the Raman frequency shift of the probe field. From the selection rules and the conservation of angular momentum, it follows that the Raman sideband of a circularly polarized probe is also circularly polarized, but with an opposite handedness. Due to this change of polarization, the strong background of the input probe light can be efficiently suppressed by means of a circular analyzer, orthogonal to the input circular polarizer (CA and CP, respectively, in Fig.4). Raman spectra of the probe pulses scattered from the rotating molecules have been measured as a function of the probe delay relative to the centrifuge with a spectrometer equipped with a 2400 grooves/mm grating.
The experimentally measured Raman spectrograms are shown in Fig.5. The bright tilted line on the left side of each plot reflects the accelerated rotation of molecules inside the centrifuge (in all three cases, the angular acceleration is exactly the same, whereas the tilt appears different due to the difference in vertical scales). While spinning up, the molecules are “leaking” from the centrifuge, producing an oscillating Raman signal in the broad range of rotational frequencies. The observed periodic oscillations indicate coherent evolution of the excited rotational wave packets, with each Raman peak corresponding to the centrifuge-induced molecular alignment Korobenko, Milner, and Milner (2014).
One can see that the oscillation period remains relatively constant at lower rotation frequencies and then grows quickly as the frequency approaches critical values, specific to each particular molecule. Beyond this frequency, the rotational Raman signal disappears altogether, pointing to the fact that a molecule can no longer follow the centrifuge field, either because Coriolis force makes it fall behind the rotating trap ( and ) or because of the rotation-induced dissociation (). Supporting this conclusion is the observation of the Raman signal merging into a single line as the revival period diverges at the Coriolis wall (mostly pronounced in the spectrogram), which suggests that the rotational levels of and become equidistant. In contrast to this behavior, the revival period of remains finite even as the molecule falls out of the centrifuge due to dissociation.
The results of the quantitative analysis of are shown in Fig.6. At lower frequencies, the experimental revival periods follow closely the dependencies derived from Eq.5 with determined as the energy minimum of the corresponding Morse-cosine potential energy surface combined with the rotational term (Eq.3). In this regime of negligible centrifugal distortion, the revival period is simply for asymmetric and molecules, and for symmetric , where is the corresponding rotational constant and is the speed of light in vacuum. The difference stems from the opposite contribution of the rotational wave packets with all-odd and all-even values, separately created by the centrifuge in an asymmetric molecule, to the Raman signal at . On the contrary, the inversion symmetry and the nuclear spin statistics of results in the absence of odd ’s in its rotational spectrum, and hence the appearance of fractional revivals with twice shorter period.
As the rotational frequency increases towards the upper frequency limits (dashed vertical lines), the experimentally measured revival periods begin to deviate from the calculated values. We attribute the disagreement to the zero-point stretching and bending, which have been neglected in our calculations. At extremely high levels of rotational excitation, the PES of a super-rotor becomes quite distorted and zero-point vibrations have to be taken into account for the correct calculation of the equilibrium bond lengths. Work is underway to verify whether this is the case, or whether Morse-cosine expansion of higher order is required in order to explain the observed molecular dynamics.
In summary, we presented a method of probing molecular potential energy surfaces with an optical centrifuge. Our approach is complementary to that based on vibrational spectroscopy, and offers much higher excitation energies. The method has been demonstrated with three linear molecules, , and . Ultra-fast rotation, induced by the centrifuge, caused the molecular bonds to stretch by as much as 32%. In the case of , our results suggest that the molecule dissociates after climbing to the extreme rotational level with a calculated angular momentum . On the other hand, we confirmed experimentally that breaking molecular bonds with the centrifuge may not always be efficient. Incidentally, both and cannot follow the accelerated rotation of the centrifuge field beyond the respective critical values and , which fall short of the dissociation limits for each molecule. Similar analysis proved successful in describing the dynamics of centrifuged nonlinear sulfur dioxide Korobenko and Milner (2016), and can be applied to any molecule amenable to centrifuge spinning, with an ultimate goal of selective bond breaking Hasbani et al. (2002).
This research has been supported by the grants from CFI, BCKDF and NSERC and carried out under the auspices of the UBC Center for Research on Ultra-Cold Systems (CRUCS).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Green, Moore, and Polik (1992) W. H. Green, C. B. Moore, and W. F. Polik, Annual Review of Physical Chemistry 43 , 591 (1992).
- 2Polanyi and Zewail (1995) J. C. Polanyi and A. H. Zewail, Accounts of Chemical Research 28 , 119 (1995).
- 3Zúñiga et al. (1999) J. Zúñiga, J., M. Alacid, A. Bastida, F. J. Carvajal, and A. Requena, The Journal of Chemical Physics 110 , 6339 (1999).
- 4Zúñiga et al. (2000) J. Zúñiga, A. Bastida, M. Alacid, and A. Requena, The Journal of Chemical Physics 113 , 5695 (2000).
- 5Zúñiga et al. (2002) J. Zúñiga, A. Bastida, A. Requena, and E. L. Sibert, The Journal of Chemical Physics 116 , 7495 (2002).
- 6Jensen (1988) P. Jensen, Journal of Molecular Spectroscopy 128 , 478 (1988).
- 7Karczmarek et al. (1999) J. Karczmarek, J. Wright, P. Corkum, and M. Yu. Ivanov, Physical Review Letters 82 , 3420 (1999).
- 8Villeneuve et al. (2000) D. M. Villeneuve, S. A. Aseyev, P. Dietrich, M. Spanner, M. Yu. Ivanov, and P. B. Corkum, Physical Review Letters 85 , 542 (2000).