Hydrogenation Facilitates Proton Transfer Through Two-Dimensional Honeycomb Crystals
Yexin Feng, Ji Chen, Wei Fang, En-Ge Wang, Angelos Michaelides,, Xin-Zheng Li

TL;DR
This study demonstrates that hydrogenation of two-dimensional honeycomb crystals like graphene and h-BN significantly lowers the energy barrier for proton transfer, explaining rapid proton transport observed experimentally.
Contribution
First principles calculations reveal hydrogenation reduces proton penetration barriers in 2D materials, enabling fast proton transfer without lattice defects.
Findings
Hydrogenation lowers proton penetration barrier to less than 1 eV.
Destabilization of chemisorption states facilitates proton transfer.
Hydrogenated 2D materials enable efficient proton transport in solutions.
Abstract
Recent experiments have triggered a debate about the ability of protons to transfer through individual layers of graphene and hexagonal boron nitride (h-BN). However, calculations have shown that the barriers to proton penetration can, at more than 3 eV, be excessively high. Here, on the basis of first principles calculations, we show that the barrier for proton penetration is significantly reduced, to less than 1 eV, upon hydrogenation even in the absence of pinholes in the lattice. Analysis reveals that the barrier is reduced because hydrogenation destabilises the initial state (a deep-lying chemisorption state) and expands the honeycomb lattice through which the protons penetrate. This study offers a rationalization of the fast proton transfer observed in experiments, and highlights the ability of proton transport through single-layer materials in hydrogen rich solutions.
Click any figure to enlarge with its caption.
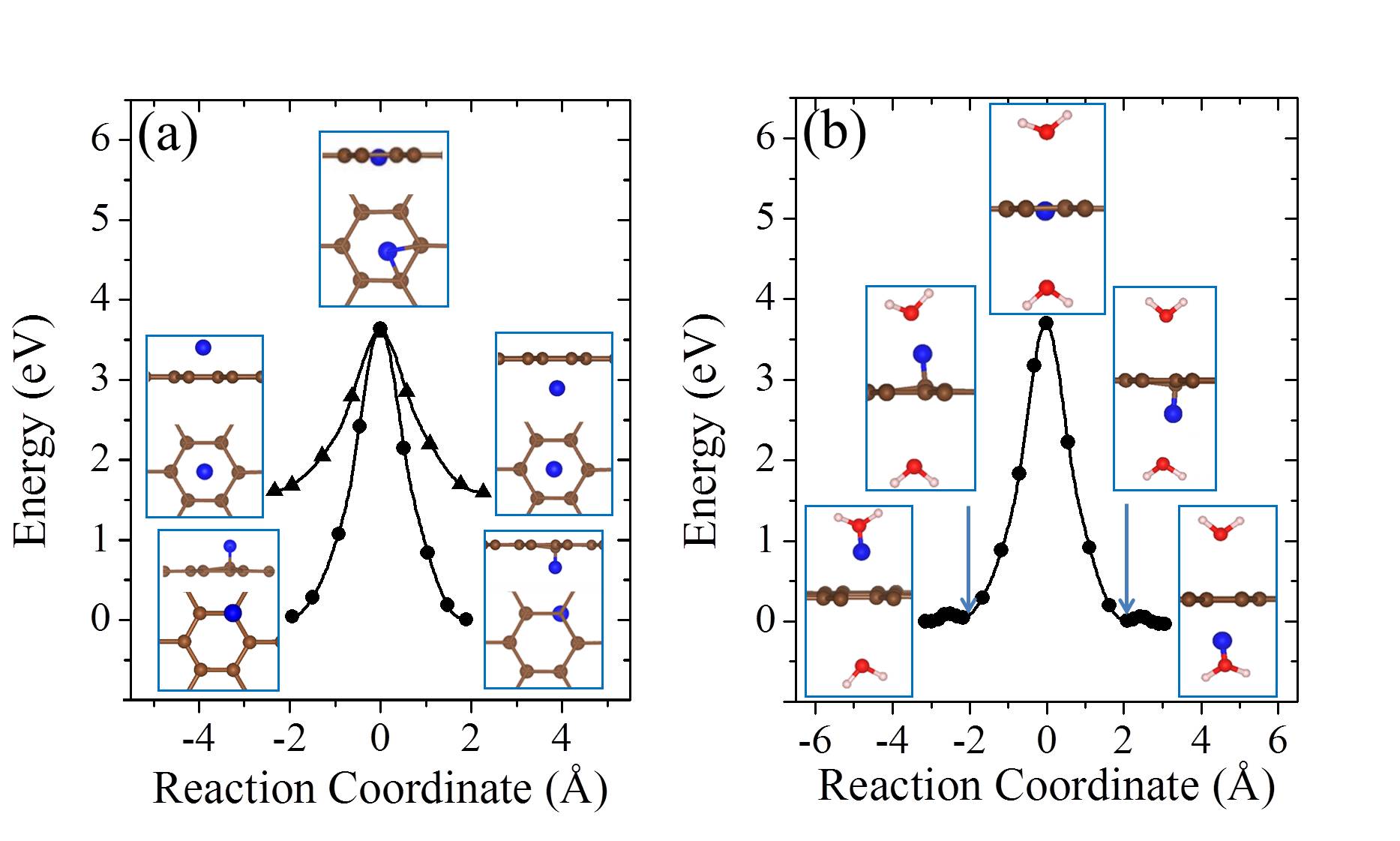 Figure 1
Figure 1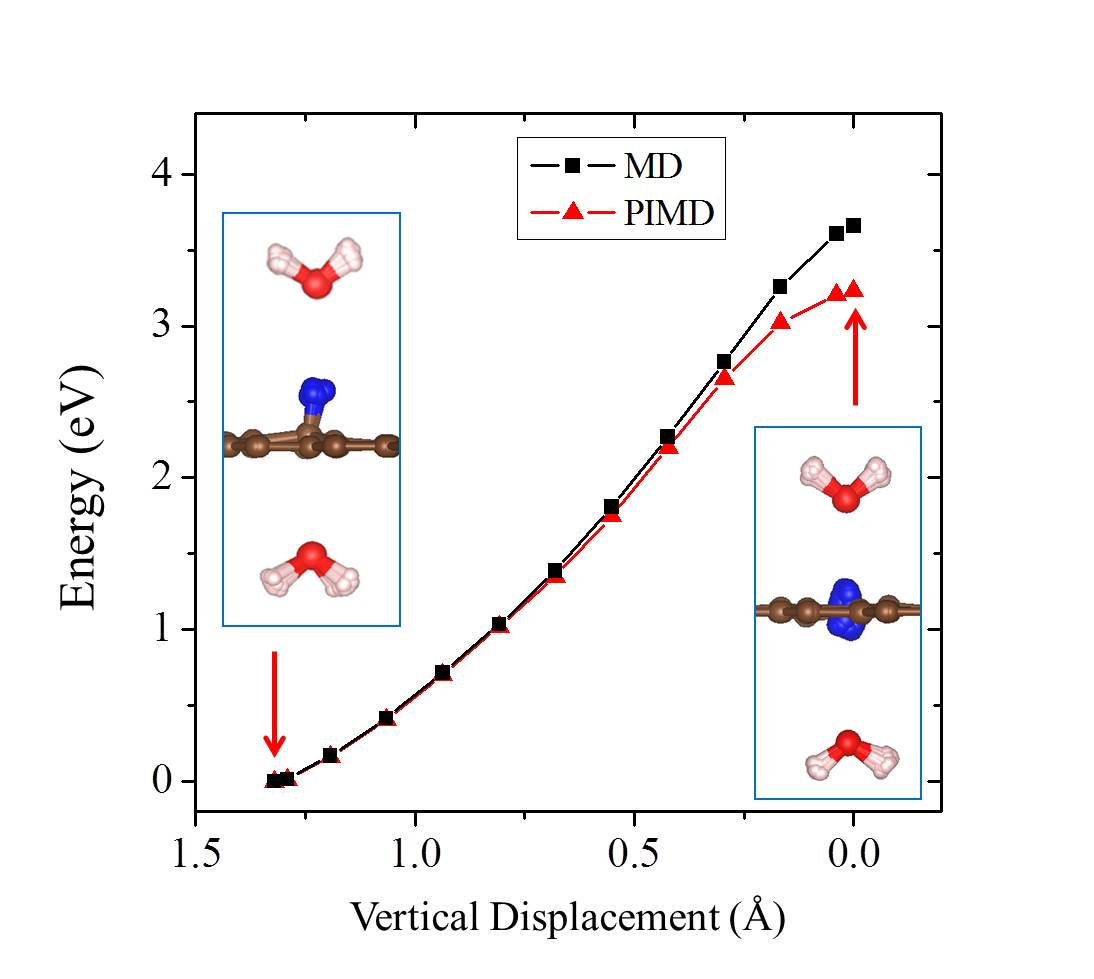 Figure 2
Figure 2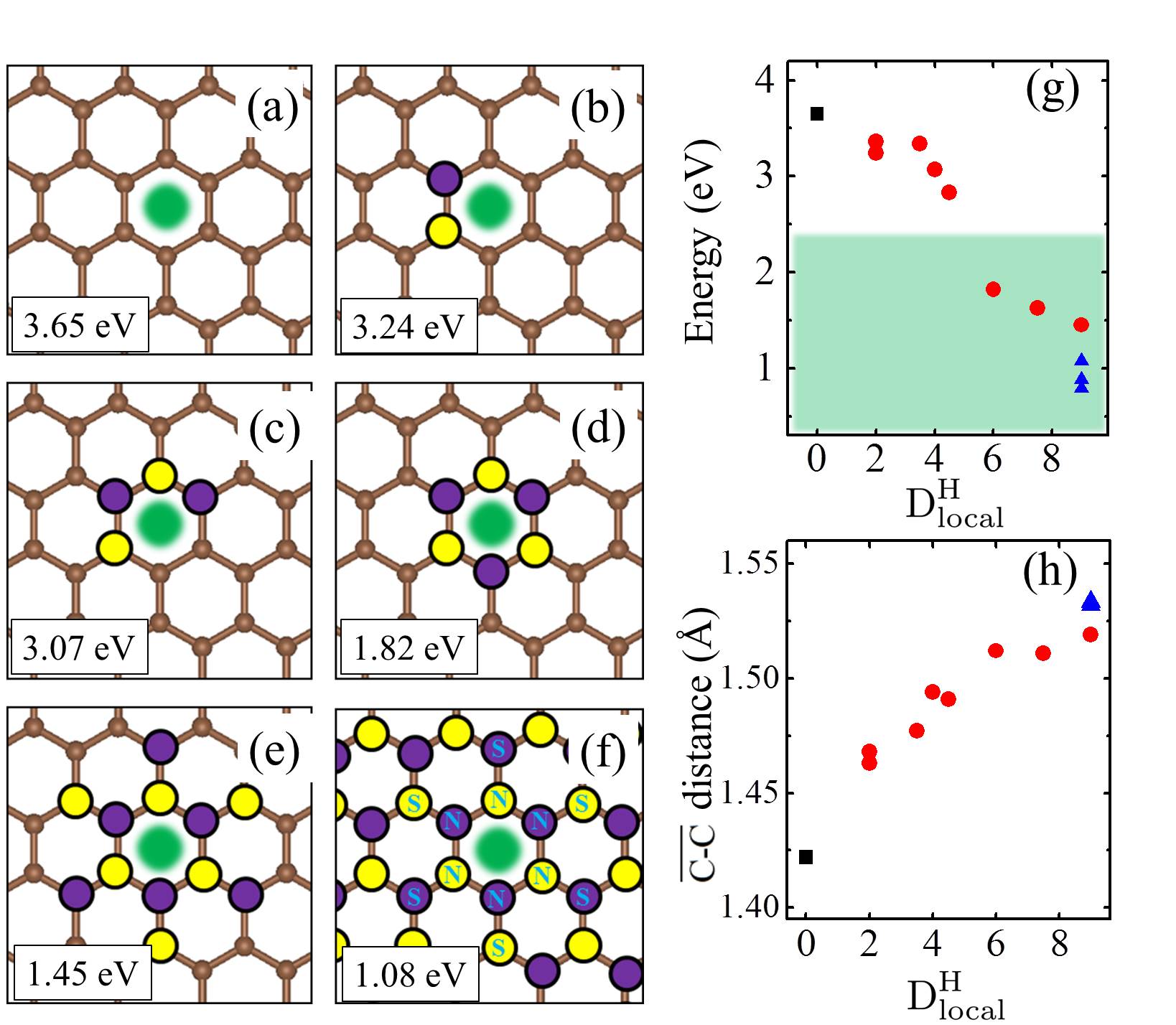 Figure 3
Figure 3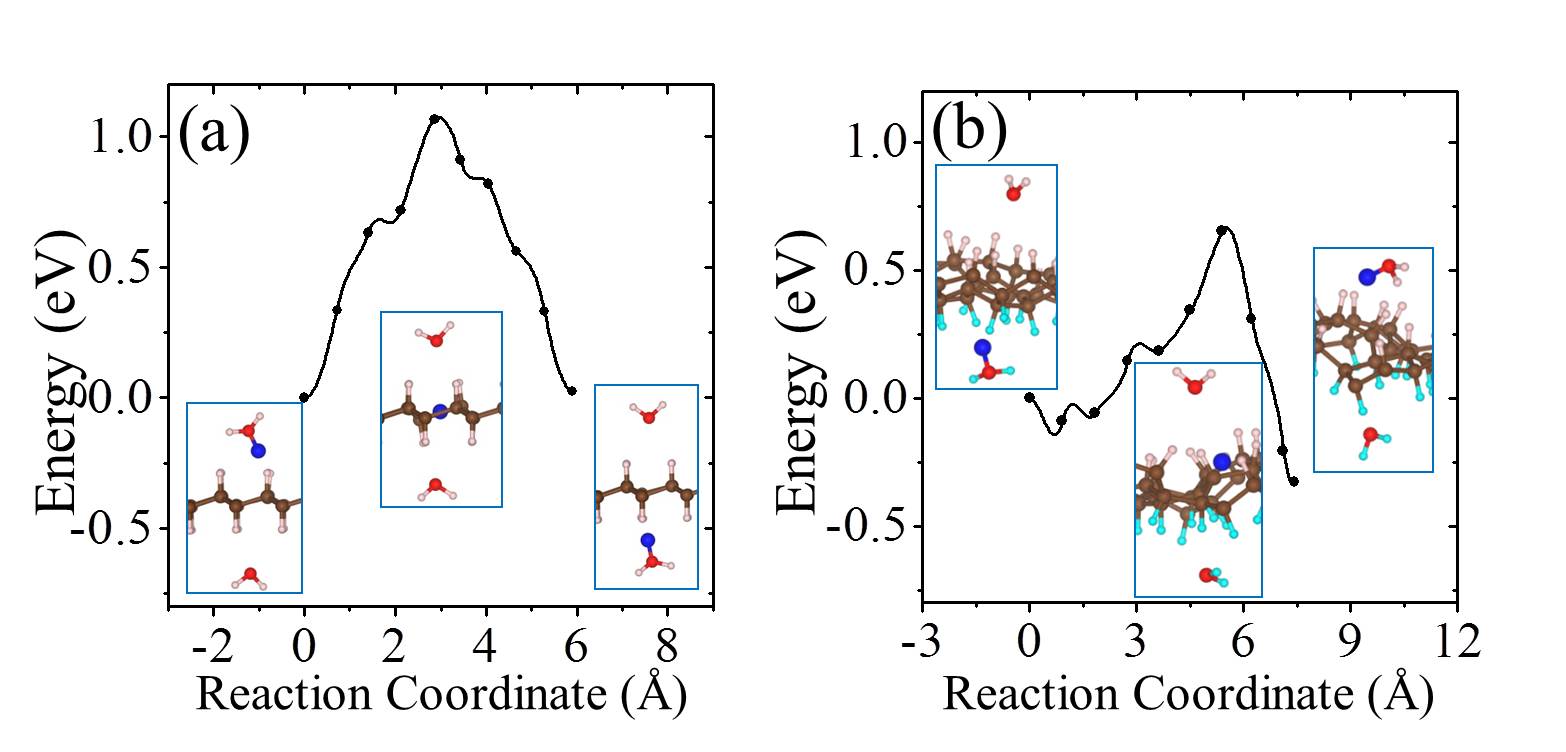 Figure 4
Figure 4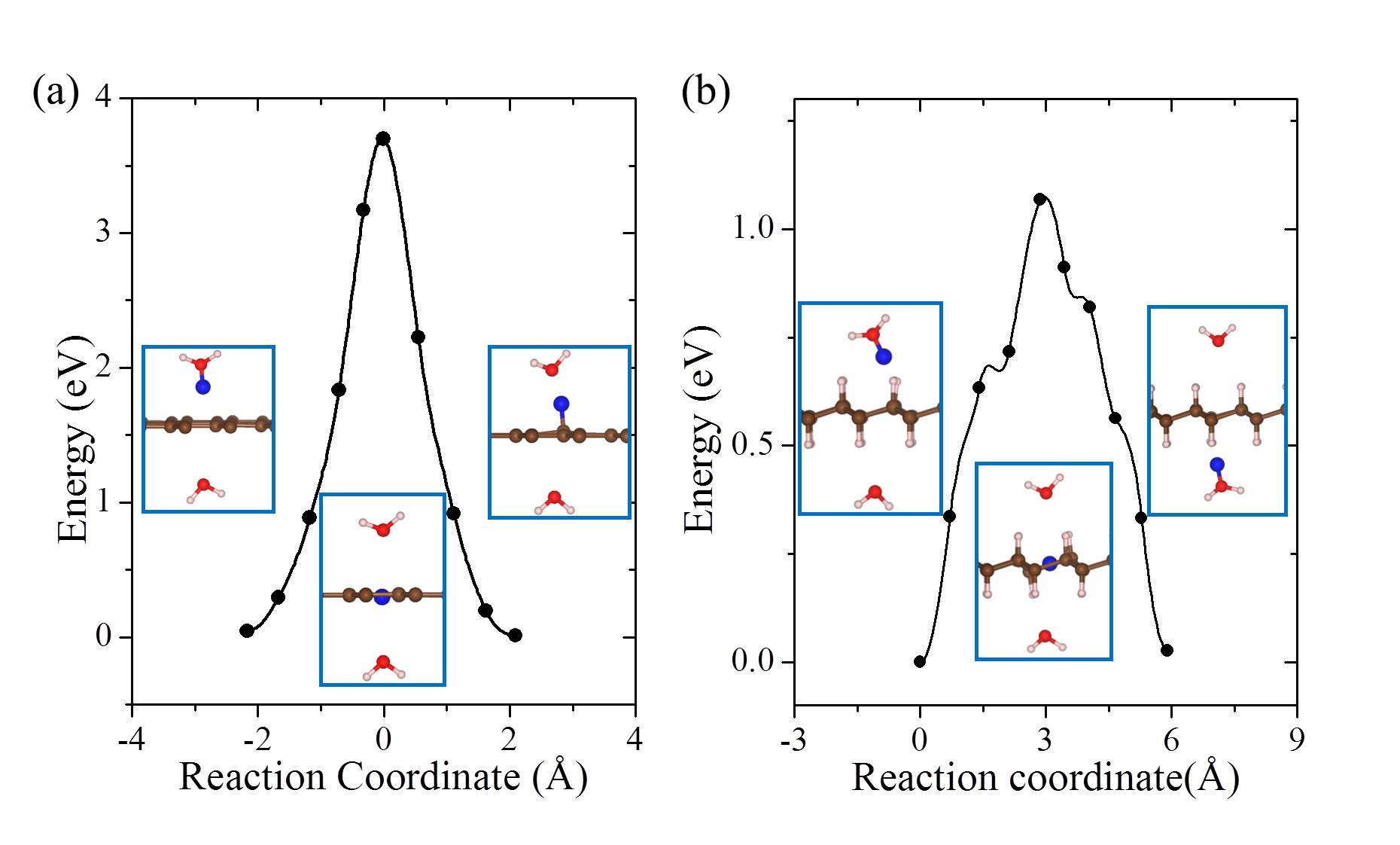 Figure 5
Figure 5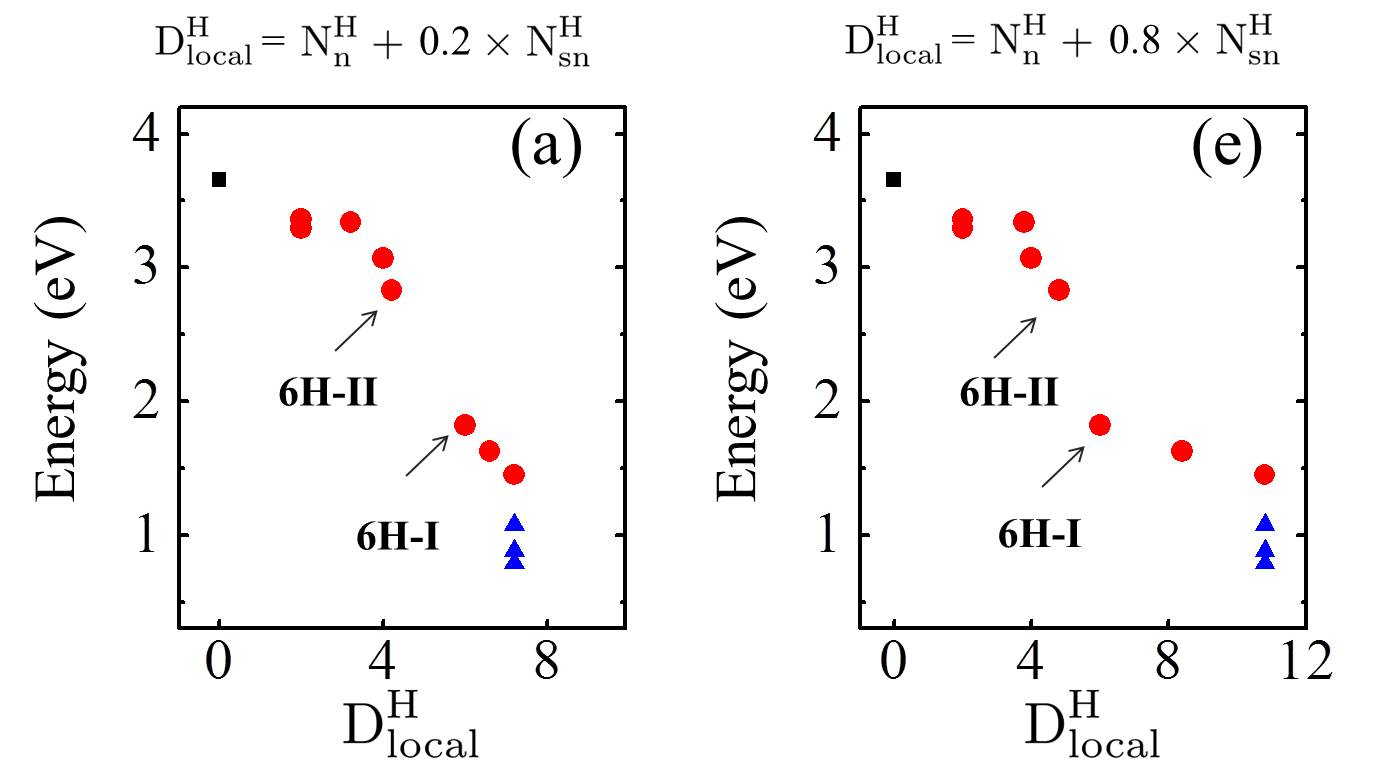 Figure 6
Figure 6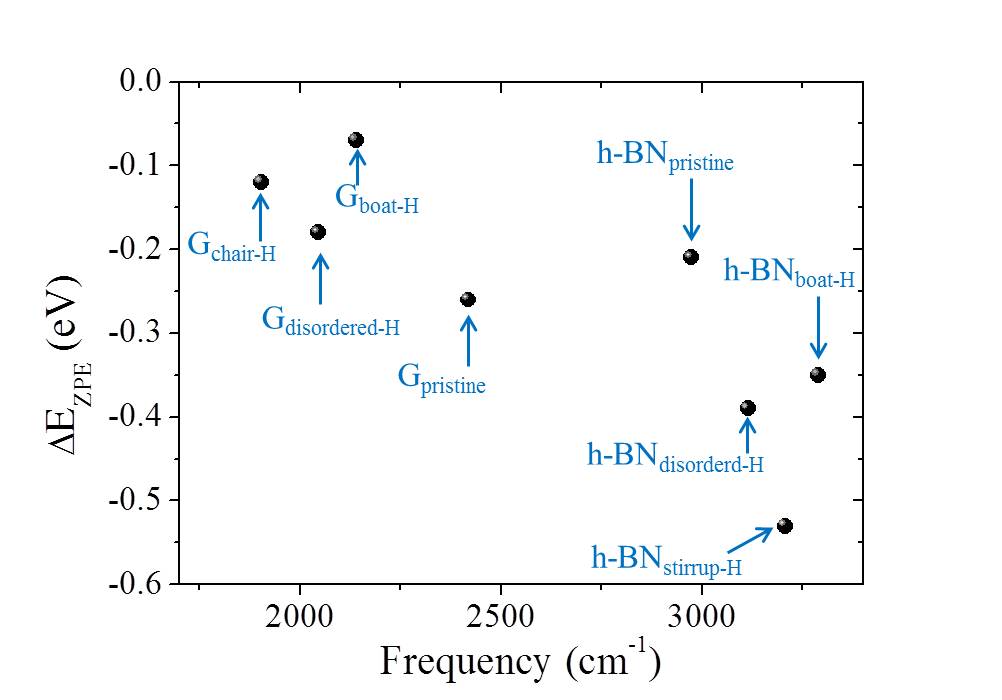 Figure 7
Figure 7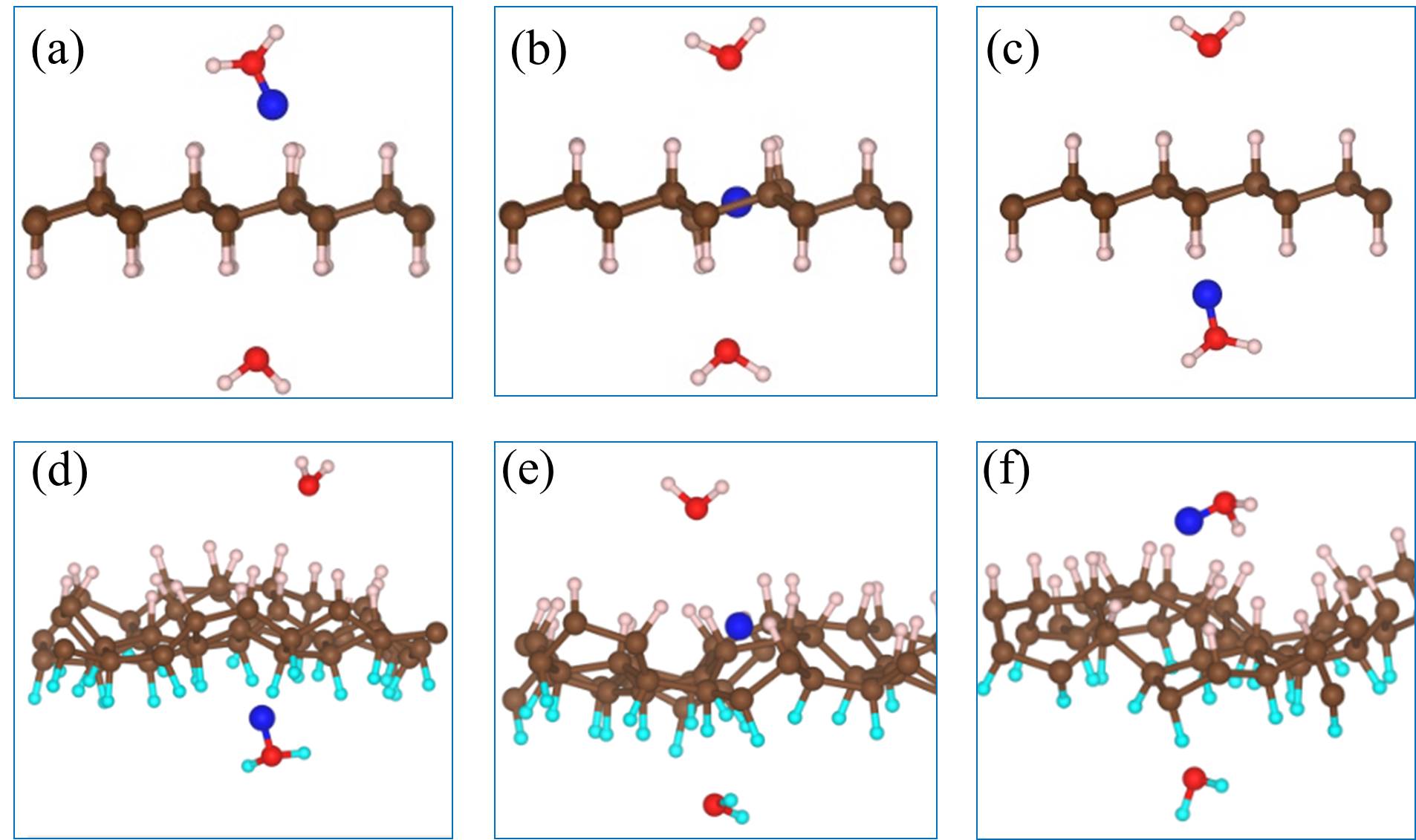 Figure 8
Figure 8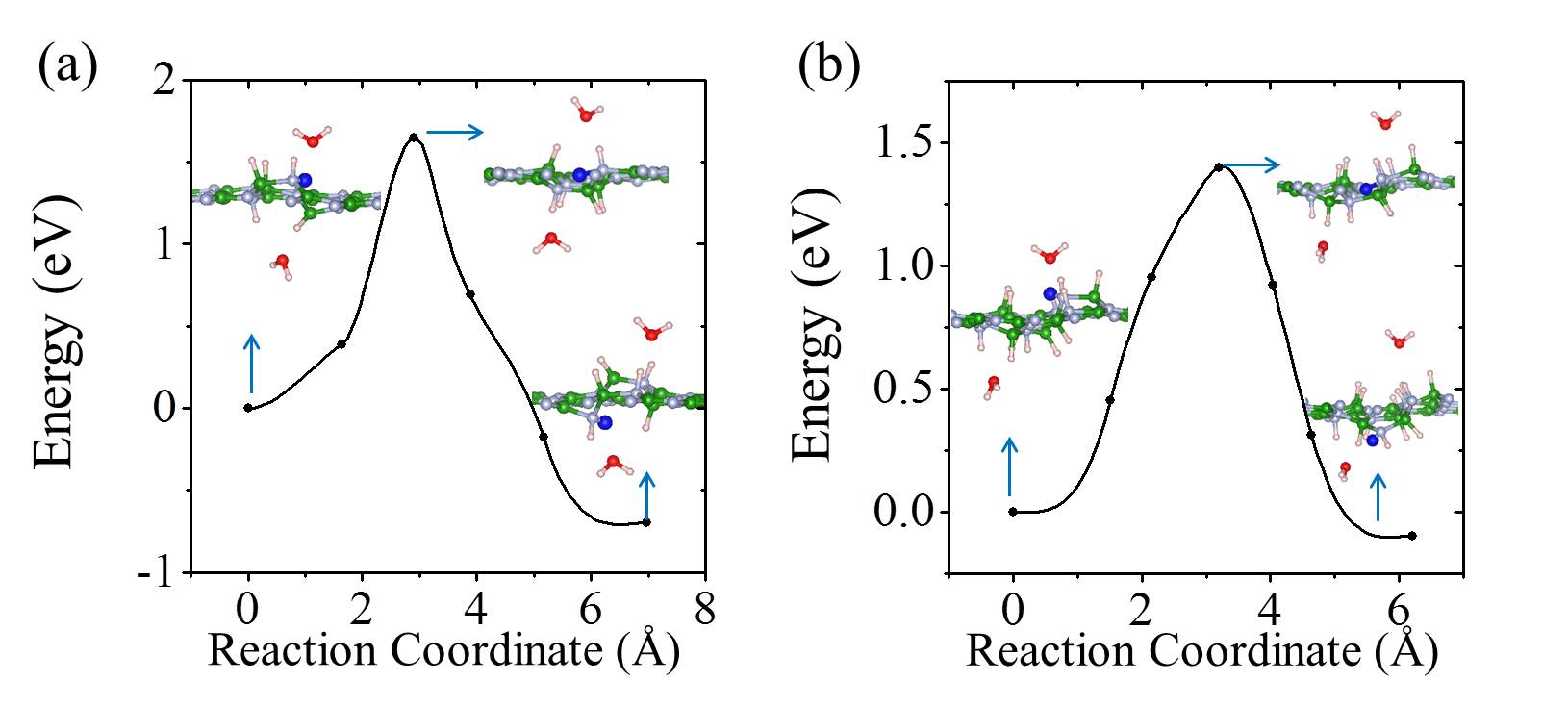 Figure 9
Figure 9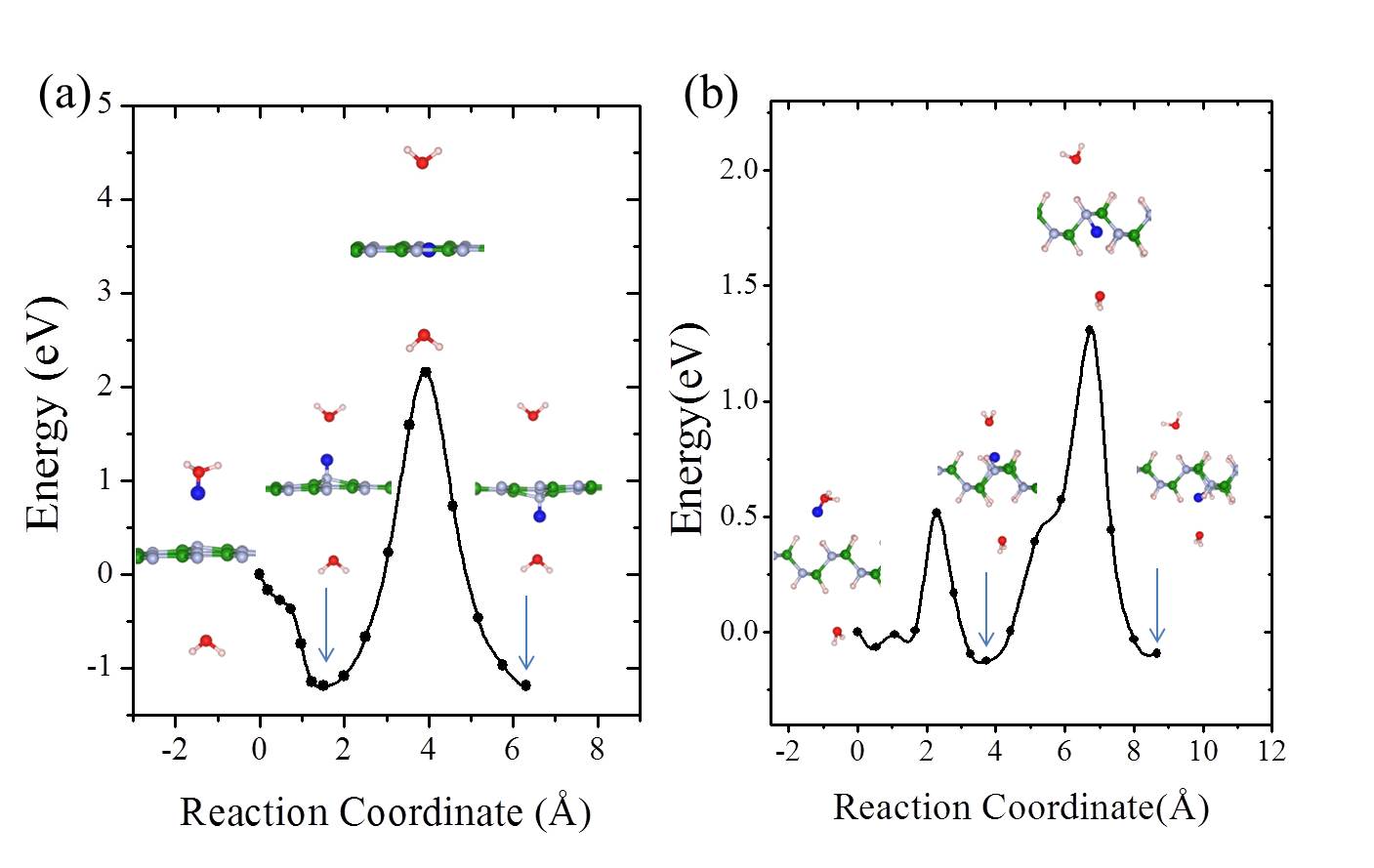 Figure 10
Figure 10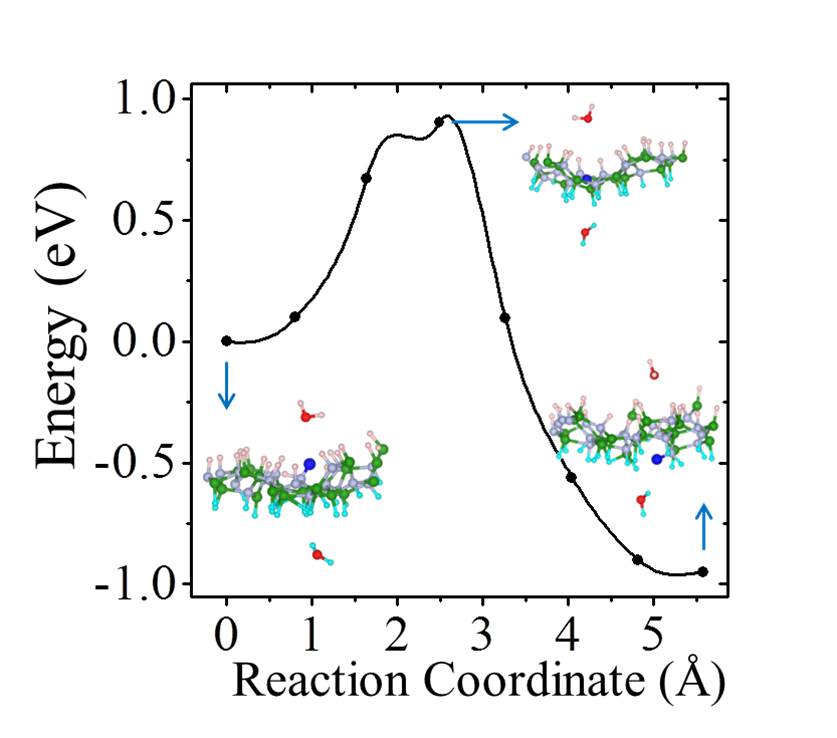 Figure 11
Figure 11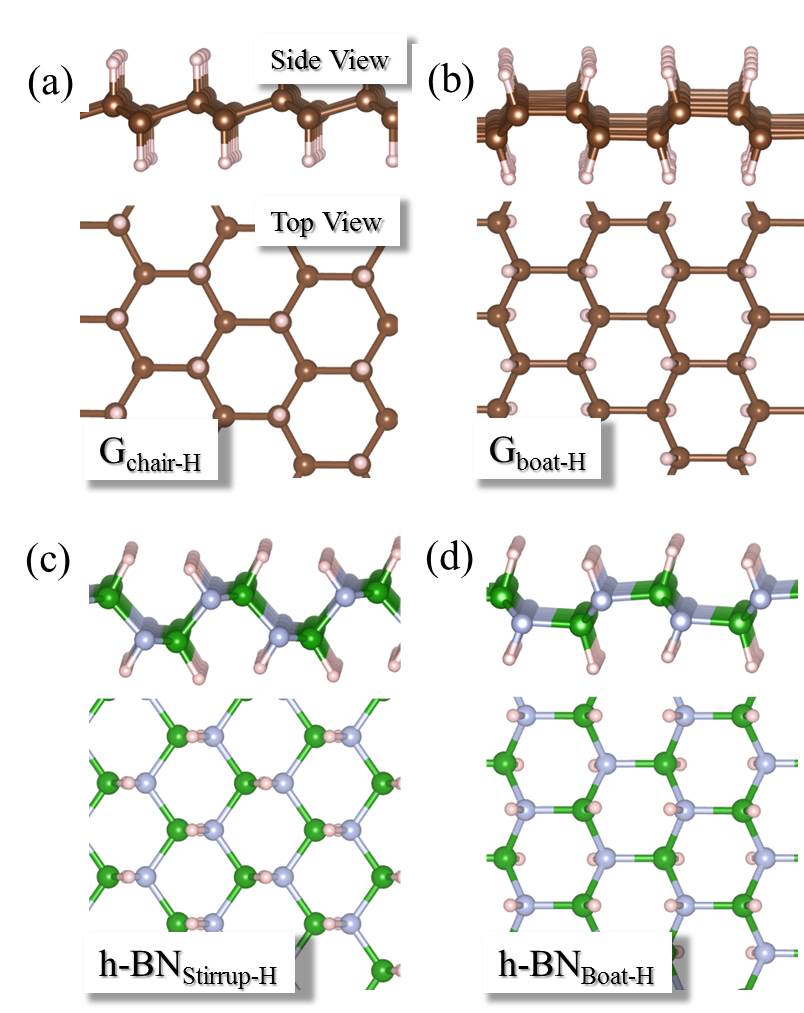 Figure 12
Figure 12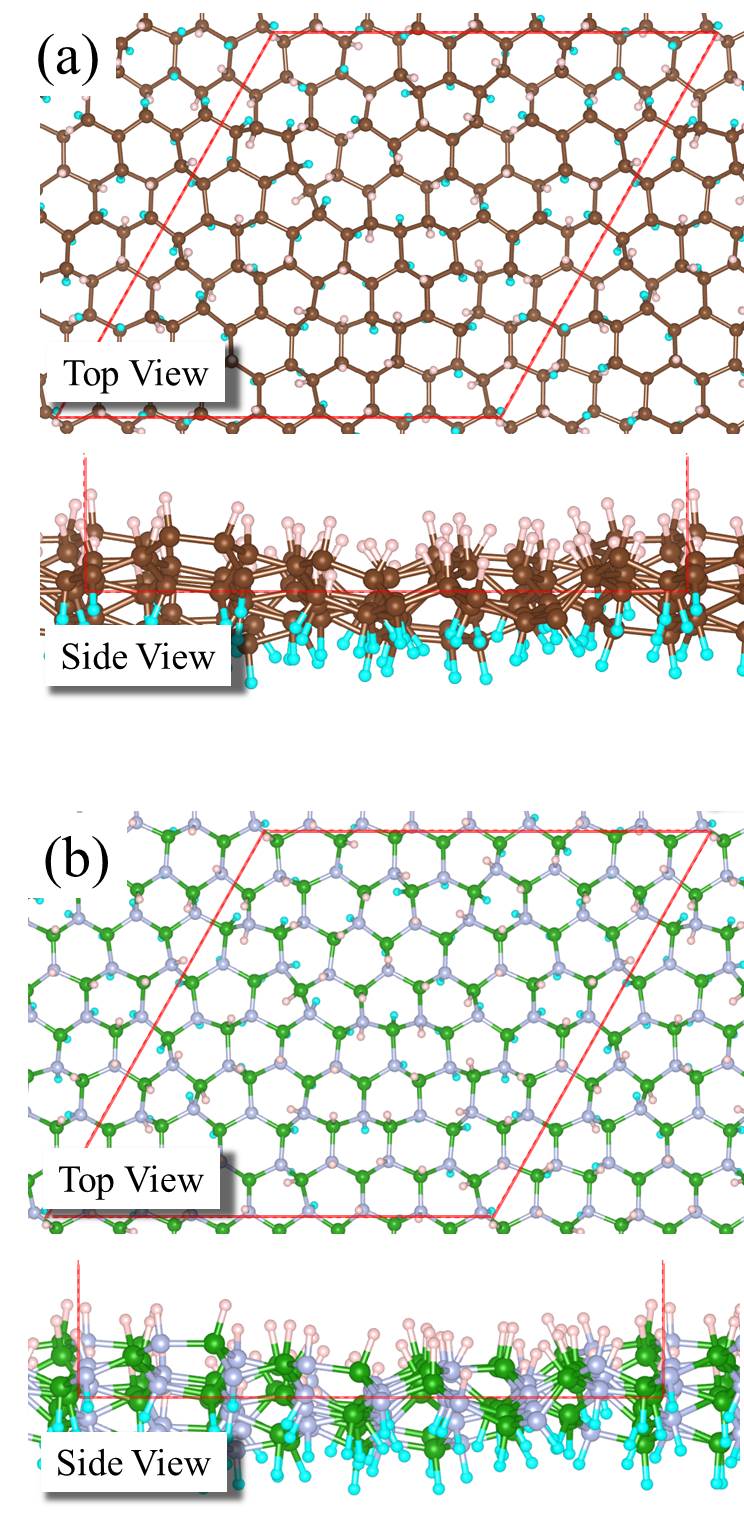 Figure 13
Figure 13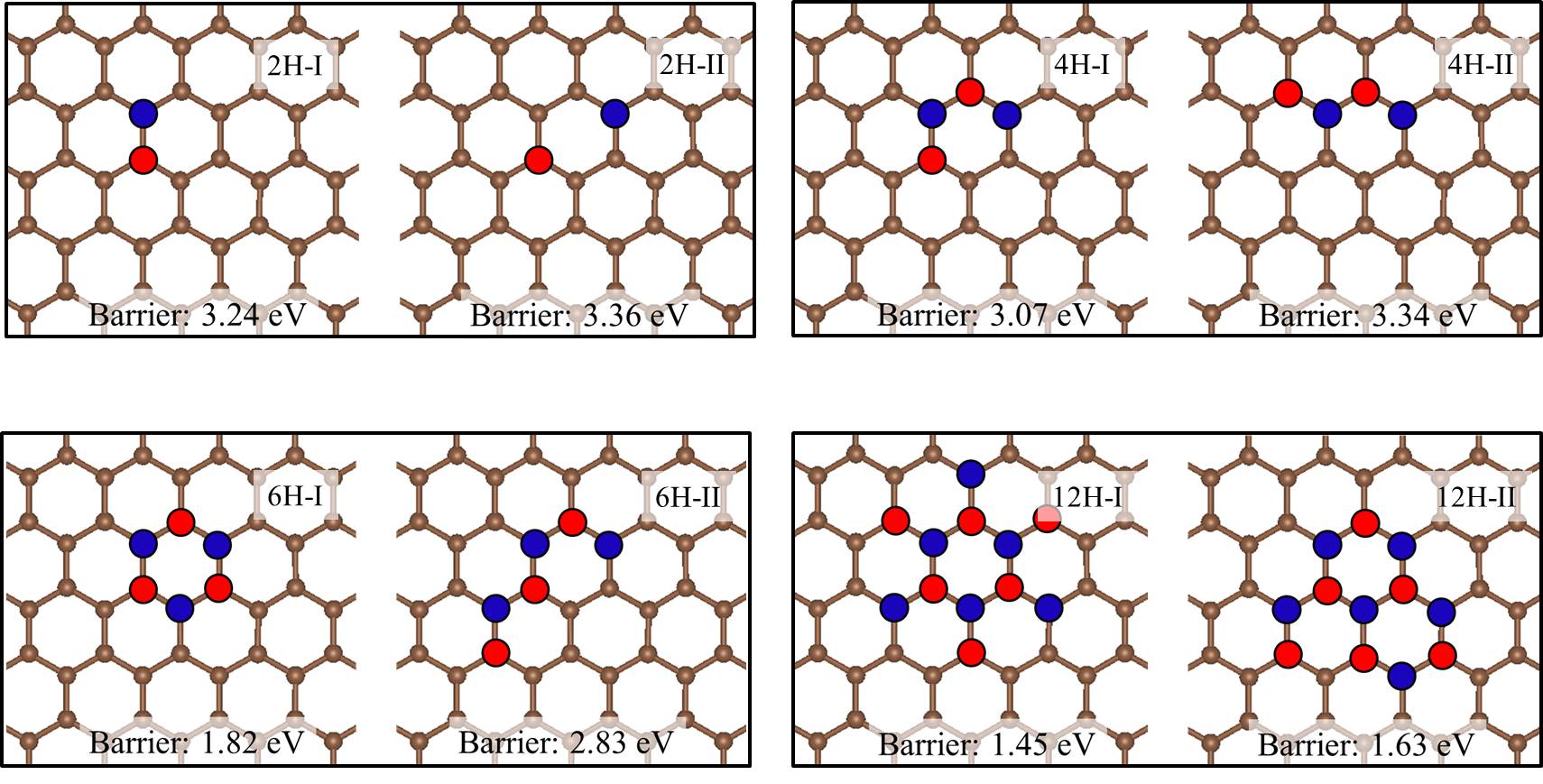 Figure 14
Figure 14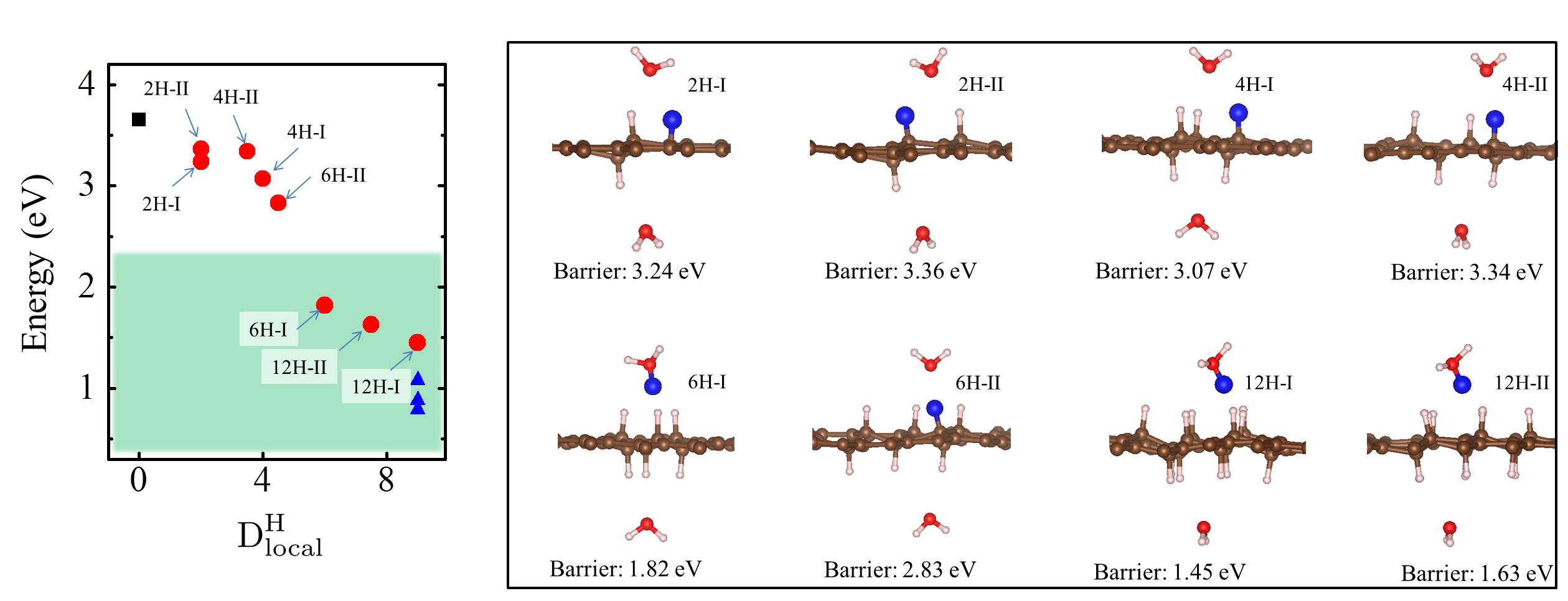 Figure 15
Figure 15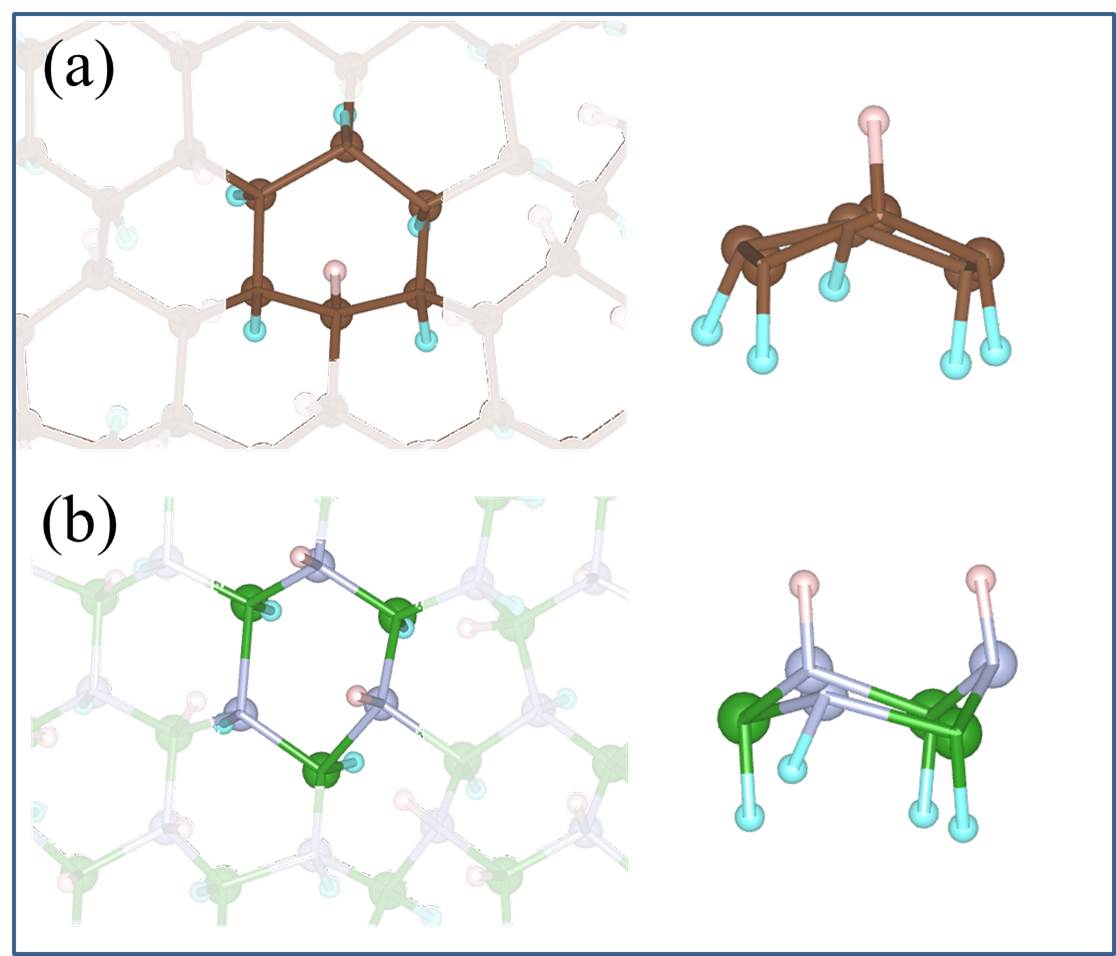 Figure 16
Figure 16 Figure 17
Figure 17 Figure 18
Figure 18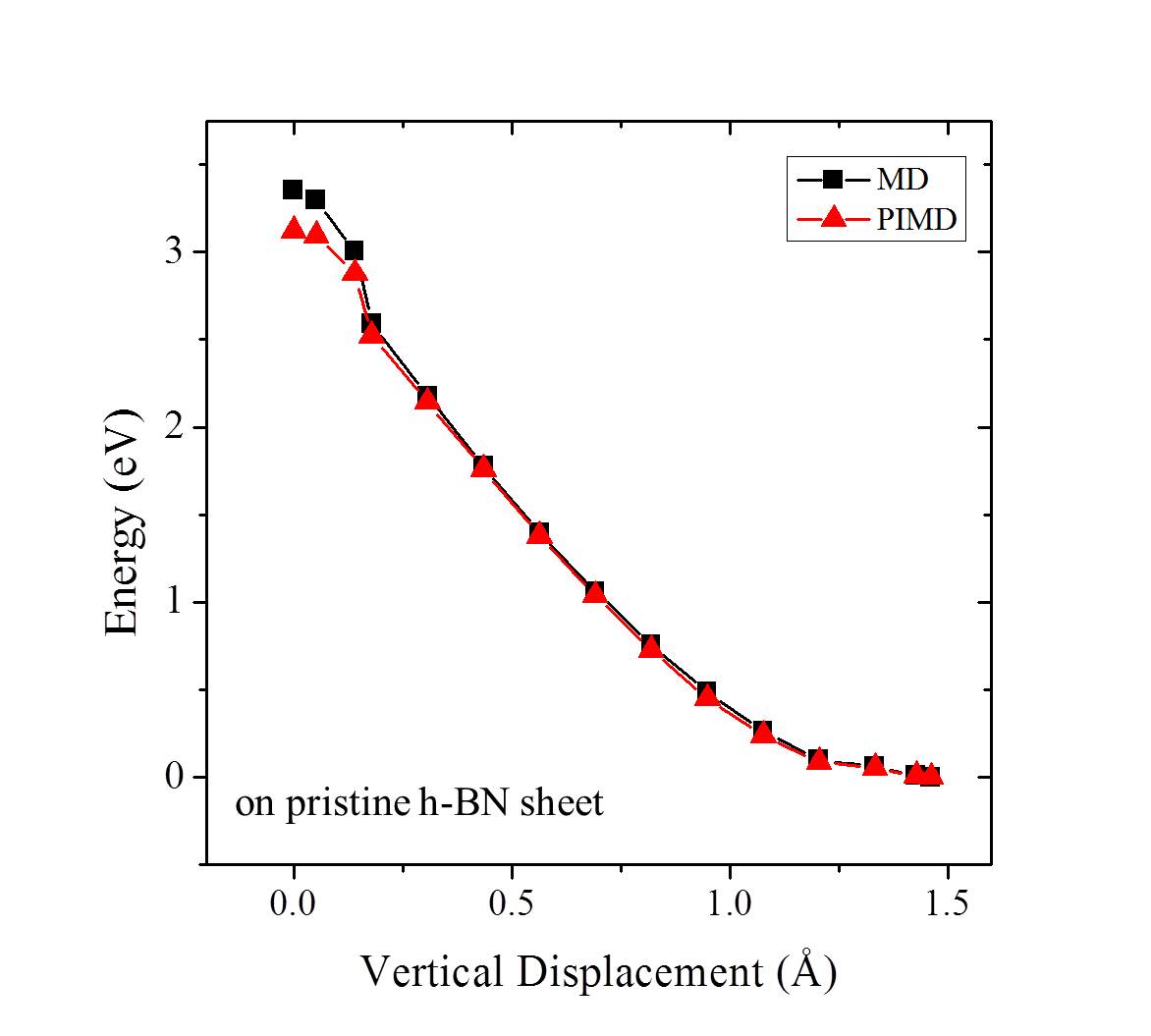 Figure 19
Figure 19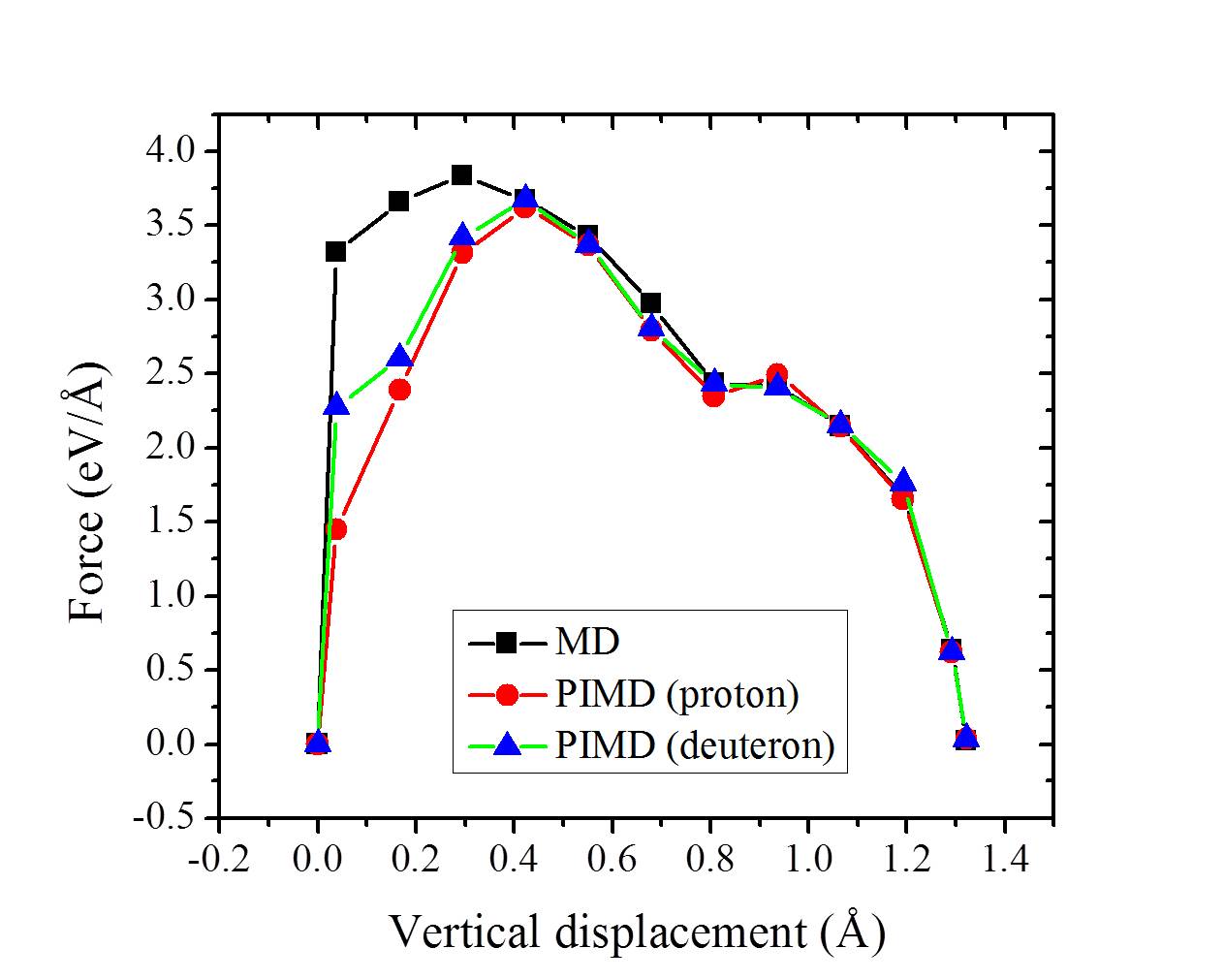 Figure 20
Figure 20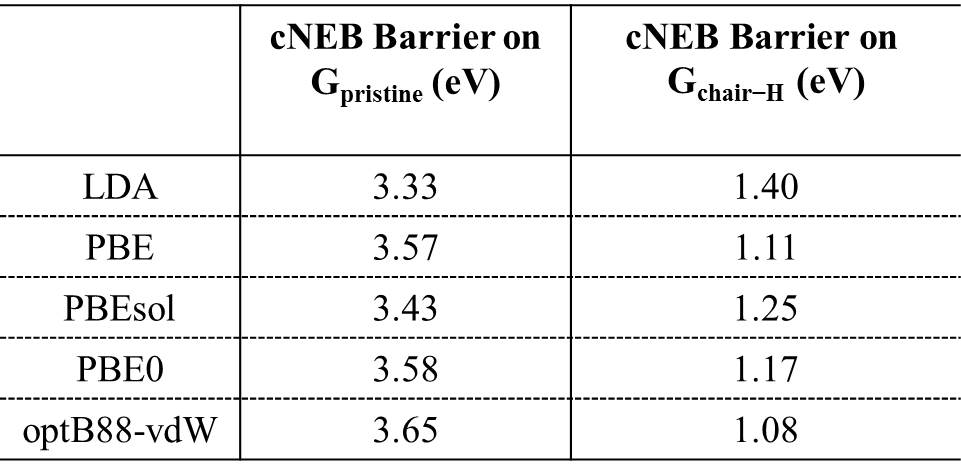 Figure 21
Figure 21| cNEB Barrier | Barrier | ||
|---|---|---|---|
| G | 3.65 | -0.26 | 3.39 |
| G | 1.08 | -0.07 | 1.06 |
| G | 0.88 | -0.12 | 0.76 |
| G | 0.79 | -0.18 | 0.61 |
| h-BN | 3.33 | -0.21 | 3.12 |
| h-BN | 1.43 | -0.53 | 0.90 |
| h-BN | 1.91 | -0.35 | 1.56 |
| h-BN | 0.93 | -0.39 | 0.51 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGraphene research and applications · Covalent Organic Framework Applications · Nanopore and Nanochannel Transport Studies
††thanks: These two authors contributed equally††thanks: These two authors contributed equally
Hydrogenation Facilitates Proton Transfer Through Two-Dimensional Honeycomb Crystals
Yexin Feng
School of Physics and Electronics, Hunan University, Changsha 410082, P. R. China
Ji Chen
Thomas Young Centre, London Centre for Nanotechnology, and Department of Physics and Astronomy, University College London, London WC1E 6BT, United Kingdom
Wei Fang
Thomas Young Centre, London Centre for Nanotechnology, and Department of Chemistry, University College London, London WC1E 6BT, United Kingdom
En-Ge Wang
School of Physics, ICQM, and Collaborative Innovation Center of Quantum Matter, Peking University, Beijing 100871, P. R. China
Angelos Michaelides
Thomas Young Centre, London Centre for Nanotechnology, and Department of Physics and Astronomy, University College London, London WC1E 6BT, United Kingdom
Xin-Zheng Li
School of Physics, ICQM, and Collaborative Innovation Center of Quantum Matter, Peking University, Beijing 100871, P. R. China
Abstract
Recent experiments have triggered a debate about the ability of protons to transfer through individual layers of graphene and hexagonal boron nitride (h-BN). However, calculations have shown that the barriers to proton penetration can, at more than 3 eV, be excessively high. Here, on the basis of first principles calculations, we show that the barrier for proton penetration is significantly reduced, to less than 1 eV, upon hydrogenation even in the absence of pinholes in the lattice. Analysis reveals that the barrier is reduced because hydrogenation destabilises the initial state (a deep-lying chemisorption state) and expands the honeycomb lattice through which the protons penetrate. This study offers a rationalization of the fast proton transfer observed in experiments, and highlights the ability of proton transport through single-layer materials in hydrogen rich solutions.
pacs:
68.43.Bc, 82.45.Mp, 71.15.Pd, 82.65.+r
Selective sieving of ions and molecules through thin membranes is a key step for a wide range of applications such as water purification and ion exchange membrane fuel cells Geim2014 ; Geim2016 ; Graphene-oxide-Manchester ; Surwade2015 ; Cohen-Tanugi2012 ; Zilman2009 ; OHern2012 ; Celebi2014 ; Achtyl2015 ; OHern2016 ; Joshi2014 ; Mi2014 ; Kim2013science ; ZengXC2015 ; Cheng2016SciAdv ; Bocquet2016 . Two-dimensional (2D) materials like graphene and hexagonal boron nitride (h-BN) offer potential as membrane materials since they are a single atom thick and have high mechanical stability and flexibility Geim2014 ; Geim2016 ; Surwade2015 ; Cohen-Tanugi2012 ; OHern2012 ; Celebi2014 ; Achtyl2015 ; OHern2016 . For some time, it was believed that pristine graphene and h-BN were impermeable to ions due to high energy barriers for penetration Miao2013 ; NJP2010 . Recent experiments, however, have suggested that protons can in fact penetrate pristine graphene and h-BN Geim2014 ; Geim2016 . In the measurements, the 2D materials were immersed in proton conducting polymers or aqueous solutions, and from temperature ()-dependent proton conductivity measurements, proton penetration barriers of only 0.8 and 0.3 eV were estimated for single-layer graphene and h-BN, respectively Geim2014 . Note that these estimated barriers include contributions from zero-point energy (ZPE) Geim2016 . Defects such as atomic pinholes are known to facilitate proton transfer Achtyl2015 . A certain level of defects will inevitably be present, associated e.g. with sp3 carbon atoms nanolett-manchester . However in Refs. Geim2014 ; Geim2016 , various measurement techniques (transmission/tunnelling electron microscopy, Raman spectroscopy and measurements of gas leakage) were used to support the assertion that the proton transfer mechanism was not facilitated by atomic defects in the membranes.
Considerable theoretical effort has been devoted towards understanding the microscopic details of how protons penetrate 2D materials Miao2013 ; NJP2010 ; Walker2015 ; Achtyl2015 ; Isotope-2016-jpcl ; H-BN-2017-pccp . It has been established on the basis of density-functional theory (DFT) calculations that the barriers to proton penetration through pristine graphene and h-BN in vacuum can be excessively high. Specifically, computed barriers of 3.5-4.0 eV have been reported for chemisorbed protons (i.e. protons that are covalently bonded to the 2D materials) to penetrate graphene NJP2010 ; Miao2013 ; Poltavsky2016 . If the protons do not chemisorb on the surface but rather penetrate the sheet via a metastable physisorption state, smaller barriers of 1.4-2.6 eV have been reported NJP2010 ; Miao2013 ; Poltavsky2016 . However, the physisorption state is only a very shallow minimum, separated from the much more stable chemisorption state by a barrier of 0.1 eV Davidson2014 . Therefore, it seems unlikely that penetration from the physisorption state is the dominant mechanism for fast proton conduction Davidson2014 ; H-BN-2017-pccp . Nonetheless this indicates that hydrogenation of graphene is facile and that graphene sheets immersed in proton conducting polymers or aqueous solutions could be hydrogenated or protonated to some extent. In addition, given the light mass of the proton, the role of nuclear quantum effects (NQEs) such as tunnelling and zero point motion could be relevant to the process, as shown e.g. through two interesting recent computational studies Poltavsky2016 ; Isotope-2016-jpcl .
In this Letter, we report a study of proton transfer through graphene and h-BN, focusing on the transmission mechanism. Consistent with earlier studies, a very high potential energy barrier of 3.6 eV is found for proton penetration of graphene via the chemisorption state. Using ab initio path-integral molecular dynamics (PIMD) PIMD1 ; PIMD2 ; PIMD3 ; Guo2016 ; Tuckerman2001 ; ZhangQ2008 ; Walker2010 , we take into account nuclear quantum effects (NQEs) and finite temperature thermal effects. We find that NQEs reduce the penetration barrier of graphene by 0.46 eV () at 300 K, which is unlikely to be responsible for the experimentally observed high transfer rate. Upon considering the role bonded atoms play on the penetration process, created here by hydrogenation of graphene and h-BN, we find that hydrogenation can reduce the penetration barriers significantly to less than 1 eV. This reduction arises because the hydrogenation induced to transformation destabilises the deep-lying chemisorption state in which the proton can get trapped on the pristine membranes. Geometrically, hydrogenation also expands the six-atom rings through which protons transfer. Analysis of the penetration barriers associated with many distinct hydrogenated membranes reveals a clear correlation between the height of the penetration barriers and the local degree of hydrogenation at the proton transfer site. Overall this work highlights the significant difference in proton penetration barriers that can be found in the vicinities of bonded atoms and helps to rationalise the facile transport of protons through single-layer materials.
Our DFT calculations were performed using the Vienna ab initio Simulation Package (VASP) vasp1996 , with an in house implementation of the ab initio constrained-centroid MD/PIMD methods Chen2013NC ; Guo2016 . The optB88-vdW functional was chosen in the electronic structure calculations so as to obtain a good description of the hydrogen (H)-bonding interactions and dispersion forces Klimes2010 ; Klimes2011 . Charged cells were employed to describe the protons in the simulations, and we confirmed that any charge states considered were correctly characterized with Bader analysis bader-charge-book ; bader-charge . We hydrogenated graphene to varying degrees without generating pinholes, using supercells ranging from 44 to 88. After hydrogenation, the supercell shape and size was allowed to change. For each partially hydrogenated structure, we have considered the two lowest energy structures following the study of Ref. H_cluster_prb . The climbing image nudged elastic band (cNEB) method was used in calculating the static penetration barriers cNEB2000 , with a force convergence criterion of 0.03 eV and all atoms were allowed to relaxed. Beyond the static description, the classical and quantum free energy profiles were obtained with constrained MD and PIMD approaches Tuckerman2001 ; ZhangQ2008 ; Walker2010 ; with the constraint applied on the vertical distance of the proton from the 2D layer. A 0.5 fs time step was used and the imaginary-time path in the PIMD simulations was sampled with 48 replicas, at a target temperature of 300 K. After thermalization, 30,000 steps (15 ps) were collected to calculate the constraint force, for each constraint point. By integrating over the constraint forces, the free energy profiles were obtained as detailed in the supplementary information (SI).
On free-standing graphene protons adsorb preferentially at the chemisorption site directly above a carbon atom (Fig. 1 (a)). From the chemisorption site, our calculations yield a proton penetration barrier of 3.60 eV. As noted, in previous experiments, the 2D layers were surrounded by proton conducting polymers or aqueous solutions Geim2014 ; Geim2016 ; Walker2015 ; Achtyl2015 ; H-BN-2017-pccp . In the current study, we do not aim to model such an aqueous environment. However, in order to gain an initial understanding of how the presence of water might impact upon the proton penetration process, we employed the simplified model shown in the inset of Fig. 1 (b). This model contains one water molecule on each side of the graphene layer and with the addition of a proton it enables us to model proton transfer from an on one side of the sheet to an on the other side of the sheet Kurita2008 . Our calculations show that the proton adsorbs at either the water molecule or the chemisorption site of the graphene sheet with very similar stability (Fig. 1 (b)). The metastable physisorption site for protons on free-standing graphene, as illustrated in Fig. 1 (a), disappears due to the presence of water. The energy barrier for a proton to transfer from the to the chemisorption site is less than 0.1 eV. The penetration barrier from the chemisorption site is 3.65 eV when water is present, very similar to the 3.60 eV obtained in the absence of water. The energy differences between physisorbed water molecules on different sites or with different orientations are only a few meV Kurita2008 ; MaJie2011 ; Hamada2012 , so different configurations of water molecules will not obviously influence the energy profile of the proton penetration process. Therefore, in agreement with recent work H-BN-2017-pccp , we conclude that the presence of water molecules is unlikely to change the fact that very high barriers exist for protons to transfer across the graphene layer.
Finite temperature and NQEs [ZPE and quantum tunneling] are known to alter the barriers of chemical processes, particularly proton transfer barriers. To understand the importance of such effects on the current system, we performed a series of ab initio MD and PIMD simulations from which free energy barriers for proton penetration were obtained. The results of these simulations are shown in Fig. 2. We find that the pure thermal effects on the barrier are relatively small and the free energy barrier in the model containing a water on either side of the sheet is 3.70 eV at 300 K. When NQEs are accounted for with PIMD the barrier is reduced by 0.46 eV at 300 K (Fig. 2), quite a substantial reduction. Analysis reveals that this reduction in the free energy barrier is due to enhanced quantum delocalisation of the proton at the transition state compared to the initial state. This is similar behavior to that observed for H chemisorption on graphene Davidson2014 , and is illustrated by the snapshots shown in Fig. 2. The reduction arising from NQEs is also in line with that reported by Poltavsky et al. when similar PIMD methods are used Poltavsky2016 , although a different computational model and reaction pathway was considered by Poltavsky et al.. However, considering the fact that the free energy barrier for the process examined remains 3 eV at 300 K, we conclude that NQEs alone can’t rationalise the experimentally observed fast proton transfer.
We noted in the introduction that carbon atoms with character are invariably present even in pristine graphene nanolett-manchester . With this in mind we explored how the presence of chemisorbed hydrogens impact the proton penetration barrier of graphene. Adsorbed hydrogens are examined since when they chemisorb they lead to an to hybridisation of the carbon atoms they are bonded to but also because the hydrogenation of graphene is facile hydrogenation-graphene . A broad range of hydrogenation scenarios was considered ranging from having just a single chemisorbed hydrogen at a proton penetration site to fully hydrogenated graphene (graphane) sheets. Examples of some of the structures considered are shown in Fig. 3, with full details given in the SI SI . Upon computing the proton penetration barriers through the various hydrogenated and partially hydrogenated sheets considered, we find that hydrogenation leads to reduced proton penetration barriers. The actual barriers obtained depends sensitively on the particular hydrogenation structure, with barriers for some hydrogenated structures reduced very substantially to 1 eV.
Let us now look more closely at the hydrogenated systems and try to understand the barriers obtained. Crucially we find that the penetration of protons through a single atom layer is a local process and that the height of the penetration barrier depends primarily on the local degree of hydrogenation in the vicinity of the penetration site. To show this more clearly, we introduce an order parameter, . is defined as , with () being the number of hydrogenated atoms at the nearest (second nearest) neighbors of the hole (indicated by N and S in the Fig. 3 (f)), and w representing a weight factor capturing the importance of hydrogenation at the second nearest sites. The barriers as a function of , with w set to 0.5, are plotted in Fig. 3 (g) (tuning w from 0.2 to 0.8 gives similar results (Fig. S11)). Upon computing for all barriers considered we found two interesting features: i) a clear correlation exists between the penetration barrier and , with the barrier getting smaller as inceases, and ii) the systems can be categorized into two main groups, with the most significant barrier reduction being found for 6. Systems with smaller than 6 belong to the group with large barriers. In these systems, the six-C ring through which the proton penetrates is not fully hydrogenated. C atoms with bonding are present in the ring and the proton can chemisorb at these sites before penetration (Figs. S4 and S5). It is the presence of the very stable chemisorption sites that lead to particularly high barriers for proton penetration. For the systems considered, when , the ring is fully hydrogenated. The bonding eliminates the deep-lying chemisorption state before penetration. In so doing the initial state energy is raised and the barriers are lower than 2.0 eV. Note that this analysis reveals that because the barrier is related to the local extent of hydrogenation, a sample does not need to have a very high global degree of hydrogenation for low barrier proton penetration sites to exist. All that is required is a high local degree of hydrogenation and indeed surface science measurements and previous calculations show that upon hydrogenation there is a tendency for Hs to cluster H_cluster_prl ; H_cluster_nanolett ; H_cluster_prb . Aside from eliminating the chemisorption well, bonded carbons also lead to an expansion of the lattice. This can be seen in Fig. 3 (h) where the averaged C-C distance of a hexagon is shown to increase with . This expansion is an additional geometric effect played by bonded carbons graphane-2010 .
Our simulations with full hydrogenation correspond to the case when the graphene layer is fully hydrogenated around a local penetration site. They have the same but different barriers in Fig. 3 (g) (three blue triangles). To understand why this happens, we take the chair conformation and a disordered H configuration as examples and show the actual cNEB barrier profiles in Fig. 4. The key difference between these two systems is that in the chair conformation, the upper and lower sides of the graphene sheet are similarly hydrogenated, while in the disordered configuration the two sides of the sheet are hydrogenated to different extents. Such asymmetric decoration creates structures wherein it is yet more facile for the proton to penetrate form one side to the other. On a larger scale, one can imagine that hydrogenation can induce different local penetration sites, with the ease of penetration related to the extent of hydrogenation on either side of the sheet.
For h-BN, H atoms also prefer to chemisorb in pairs on B and N atoms, and the averaged binding energy between H atoms and h-BN increases with the degree of hydrogenation H-BN-2016-pccp ; hBN2000 . As with graphene, upon examining proton penetration through h-BN we find that the barriers decrease upon hydrogenation. As shown in Table I, the barrier through pristine h-BN is as high as 3.33 eV. In fully hydrogenated h-BN with ordered H configurations (h-BN sheets with stirrup and boat conformations, h-BN and h-BN) H-BN-apl , the barrier can be reduced to less than 2.0 eV. For disordered H configurations, the barrier further decreases to 0.93 eV. Some representative energy profiles for partially and fully hydrogenated h-BN sheets are provided in the Figs. S14-S16.
In Table I, we summarize some representative barriers obtained for proton transfer through the various graphene and h-BN systems considered. Also included in Table I are the ZPE corrections to the barriers computed within the harmonic approximation. ZPE effects decrease the barriers to proton penetration in all systems considered and when they are taken into consideration the lowest barrier on graphene is 0.61 eV and on h-BN it is 0.51 eV. Finally, we note that we have also considered how substitution of H for D is likely to alter the penetration barriers. Treating this again at the ZPE level we find a 50 meV difference in penetration barriers between H and D for , and a 120 meV difference between H and D for . In each case, the D barrier is slightly larger than the H barrier, in agreement with recent computational work Isotope-2016-jpcl and experiment Geim2016 .
To conclude, we have reported a theoretical study on proton transfer through graphene and h-BN. After considering various factors that could impact on the penetration barriers for protons, we find that hybridization at the penetration site, achieved here through hydrogenation, plays a key role in reducing these barriers to less than 1.0 eV. The physical origin of the barrier reduction is the elimination of the deep-lying chemisorption states and the expansion of the honeycomb lattice at the penetration site. Combining the major influence from hydrogenation and minor influence from NQEs, the experimentally observed low proton transfer barrier can be rationalised. Considering the fact that 2D materials can be functionalized with various elements other than H, e.g. O, OH, F, Cl, this study suggests that there could be further scope for more controllable ion and proton sieving. We hope our study can stimulate further theoretical and experimental investigations in this direction.
Acknowledgements.
Y.X.F., X.Z.L. and E.W. are supported by the National Basic Research Programs of China under Grand Nos. 2016YFA0300900, 2013CB934600, the National Science Foundation of China under Grant Nos 11275008, 1142243, 11274012, 91021007, 11604092 and 11634001. J.C. and A.M. are supported by the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement number 616121 (HeteroIce project). A.M. is also supported by the Royal Society through a Royal Society Wolfson Research Merit Award. The computational resources were provided by the supercomputer TianHe-1A in Tianjin, China and ARCHER from the UKCP consortium (EP/F036884/1).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1(1) S. Hu et al. , Nature 516 , 227 (2014).
- 2(2) M. Lozada-Hidalgo et al. , Science 351 , 68 (2016).
- 3(3) J. Abraham et al. , Nat. Nanotech. (2017).
- 4(4) S. P. Surwade et al. , Nat. Nanotech. 10 , 459 (2015).
- 5(5) D. Cohen-Tanugi and J. C. Grossman, Nano Lett. 12 , 3602 (2012).
- 6(6) A. Zilman, J. Pearson, and G. Bel, Phys. Rev. Lett. 103 , 128103 (2009).
- 7(7) S. C. O’Hern et al. , ACS Nano 6 , 10130 (2012).
- 8(8) K. Celebi et al. , Science 344 , 289 (2014).