Triplet Fermions and Dirac Fermions in Borophene
Motohiko Ezawa

TL;DR
This paper derives a Dirac theory for borophene, revealing conditions for gapless cones, emergence of triplet fermions at three-band touching points, and analyzing edge states in nanoribbons.
Contribution
It provides an explicit Dirac theory for borophene's Dirac cones, introduces effective models for triplet fermions, and studies edge state behaviors based on edge terminations.
Findings
Dirac cones are gapless with inversion symmetry
Triplet fermions emerge at three-band touching points
Edge states vary with edge termination methods
Abstract
Borophene is a monolayer materials made of boron. A perfect planar boropehene called borophene has Dirac cones and they are well reproduced by a tight-binding model according to recent experimental and first-principles calculation results. We explicitly derive a Dirac theory for them. Dirac cones are gapless when the inversion symmetry exists, while they are gapped when it is broken. In addition, three-band touching points emerge together with pseudospin triplet fermions when all transfer energy is equal and all on-site energy is equal. The three-band touching is slightly resolved otherwise. We construct effective three-band theories for triplet fermions. We also study the edge states of borophene nanoribbons, which show various behaviors depending on the way of edge terminations.
Click any figure to enlarge with its caption.
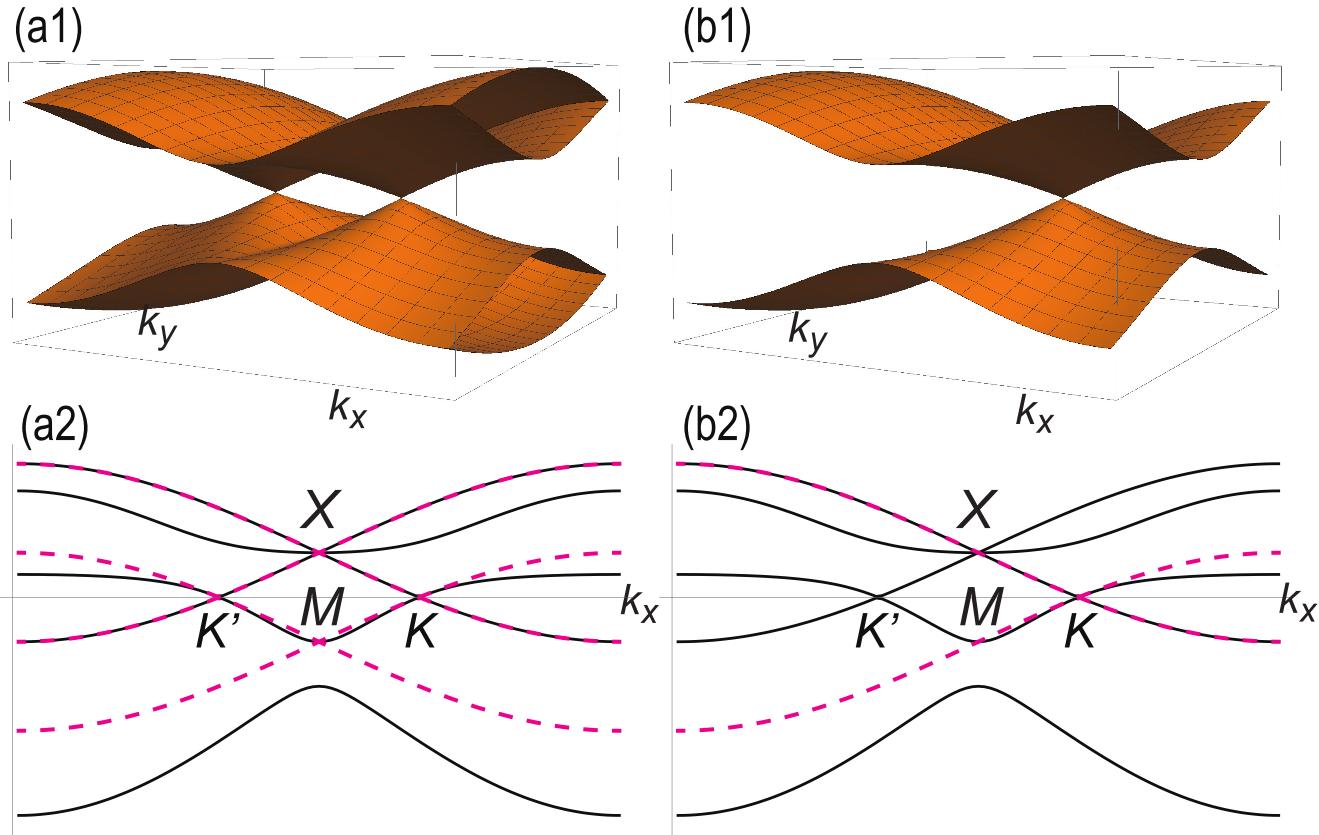 Figure 1
Figure 1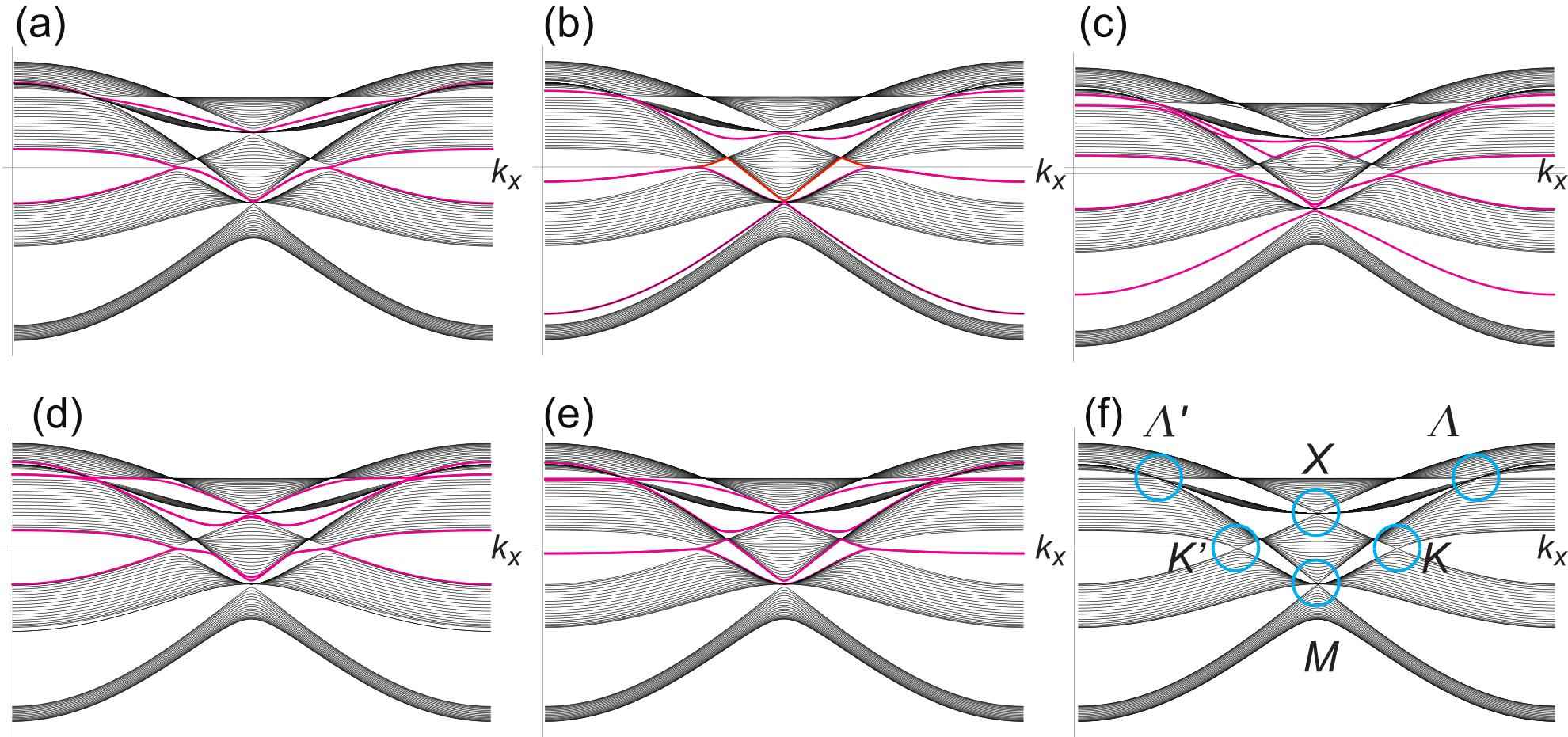 Figure 2
Figure 2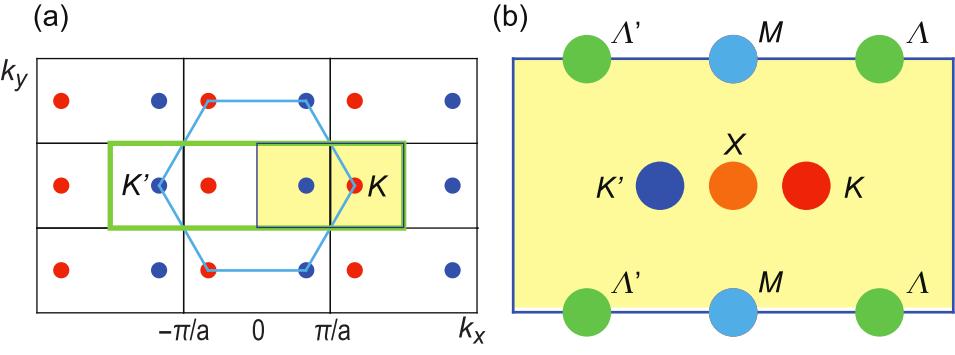 Figure 3
Figure 3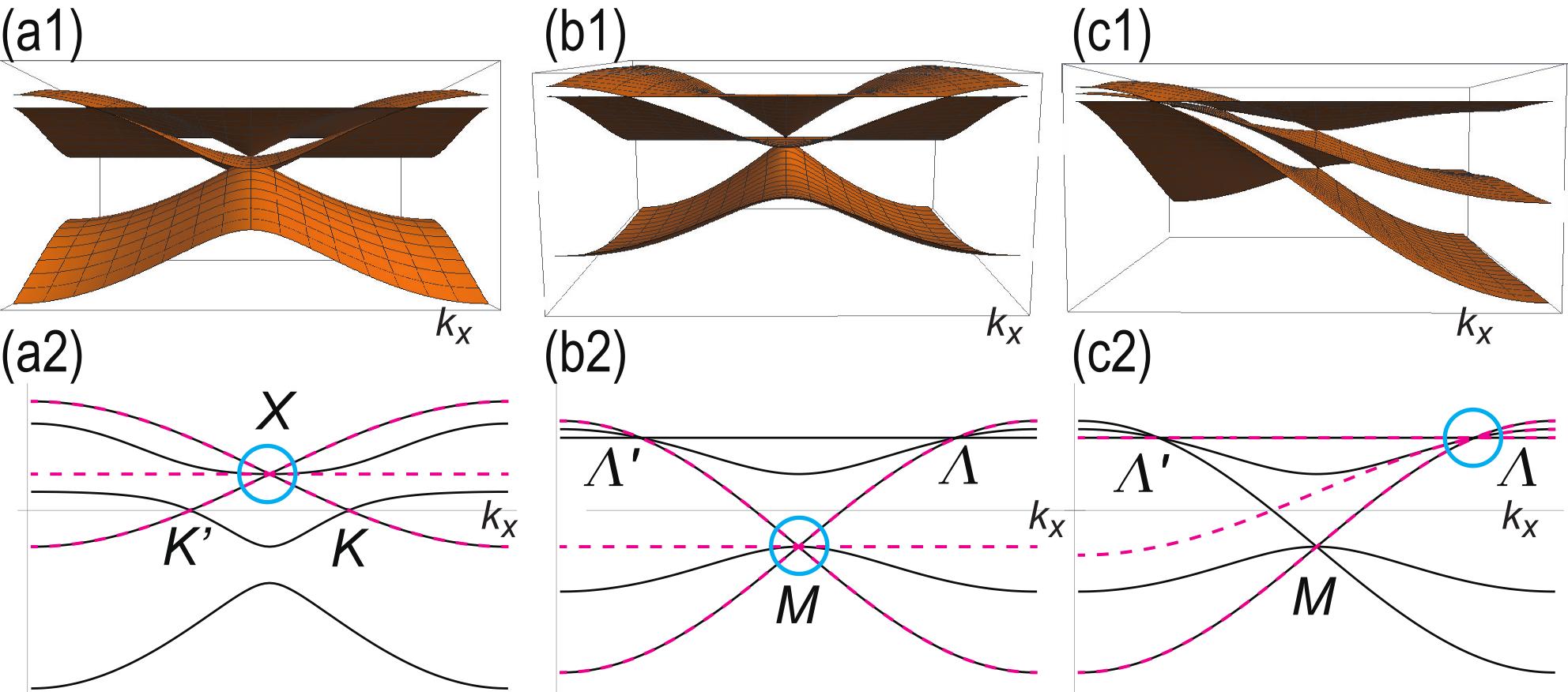 Figure 4
Figure 4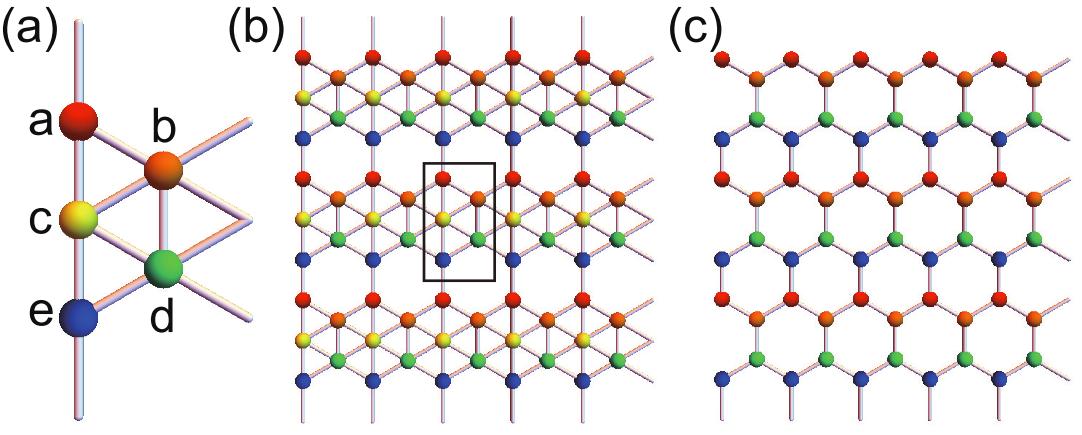 Figure 5
Figure 5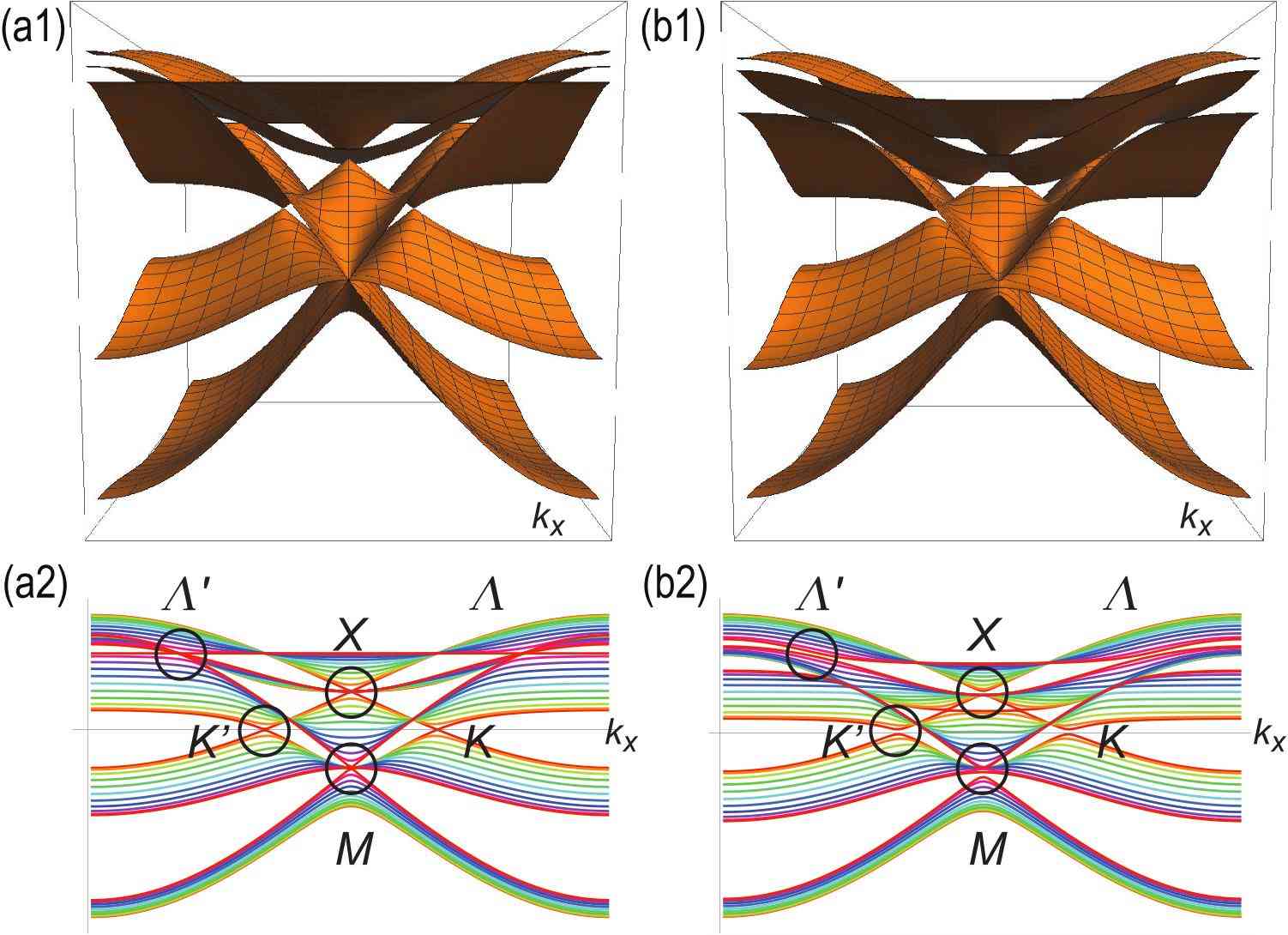 Figure 6
Figure 6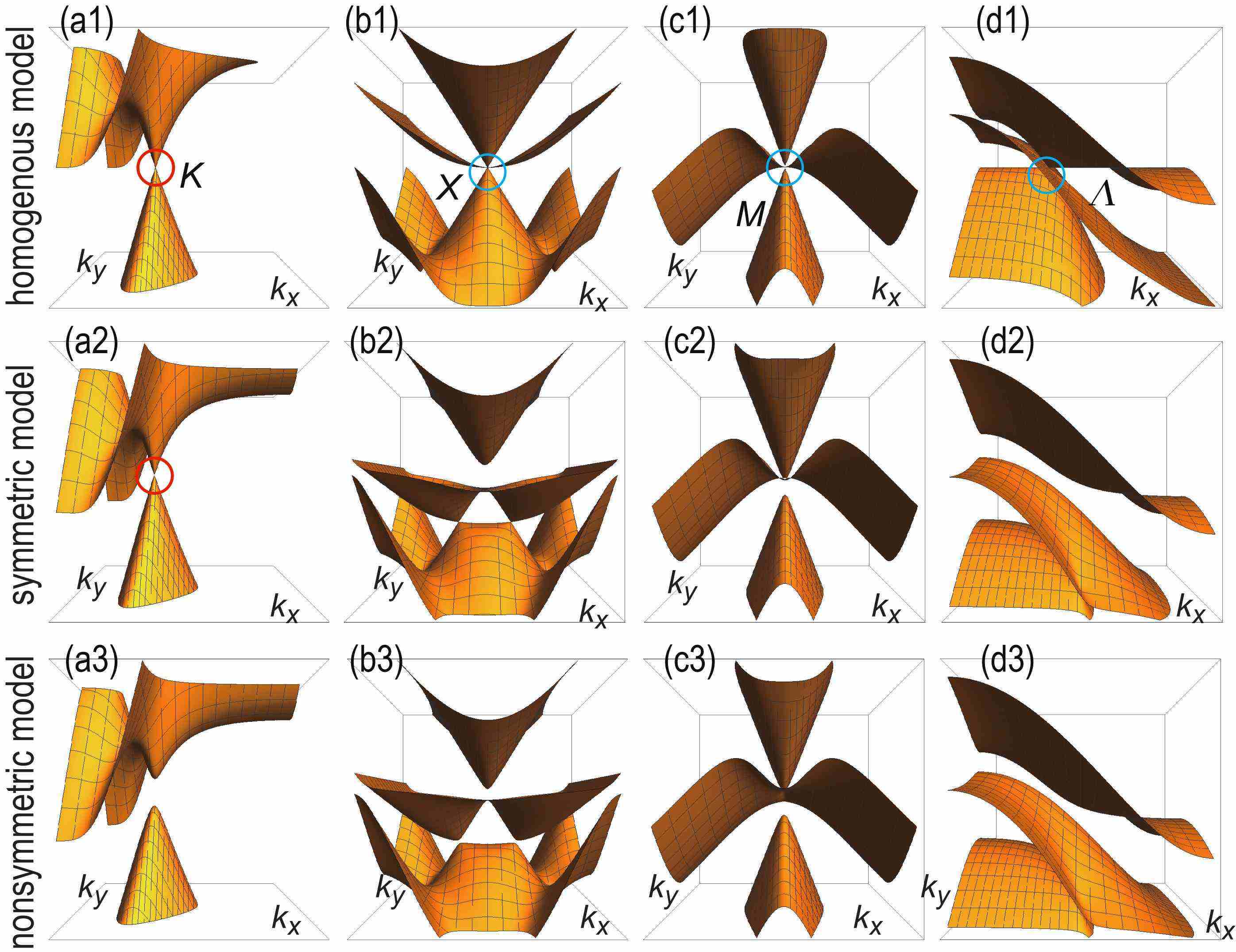 Figure 7
Figure 7Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Triplet Fermions and Dirac Fermions in Borophene
Motohiko Ezawa
Department of Applied Physics, University of Tokyo, Hongo 7-3-1, 113-8656, Japan
Abstract
Borophene is a monolayer materials made of boron. A perfect planar boropehene called borophene has Dirac cones and they are well reproduced by a tight-binding model according to recent experimental and first-principles calculation results. We explicitly derive a Dirac theory for them. Dirac cones are gapless when the inversion symmetry exists, while they are gapped when it is broken. In addition, three-band touching points emerge together with pseudospin triplet fermions when all transfer energy is equal and all on-site energy is equal. The three-band touching is slightly resolved otherwise. We construct effective three-band theories for triplet fermions. We also study the edge states of borophene nanoribbons, which show various behaviors depending on the way of edge terminations.
I Introduction
Monolayer material science is one of the most active fields of condensed matter physics in this decade. It has begun with grapheneNetoRev and been extended to the group IV monolayer materials including siliceneLiuPRB ; EzawaQAHE ; LTao , germaneneDavi ; Der ; GLi and staneneStanene . Furthermore, experimental success of phosphorenePhos1 ; Phos2 ; Phos3 has opened a field of the group V monolayer materials including arseneneArsenene and antimoneneAntimonene . A search for new monolayer materials is extended to the group III monolayer materials including borophene and alumineneAlumi . Especially, several types of borophene are proposed by first-principles calculationHTang ; XWu ; Penev ; HLiu ; ZZhang2 ; YLiu . Recently, several types of borophene is synthesized on Ag(111)Man ; ZZhang ; BFeng ; BFeng2 .
In particular, borophene experimentally manufactured on the silver surfaceBoro is quite interesting. Dirac fermions are clearly observed by the ARPES experiments as well as by first-principles calculation. The band structure is well reproduced by a tight-binding model, where it is enough to take into account only the orbitals due to its perfect planar structure. The unit cell contains five atoms as in Fig.1(a). The lattice has a perfectly flat structure as in Fig.1(b). It can be constructed by adding atoms indicated in yellow into the honeycomb lattice.
In this paper we study the band structure of borophene based on the tight-binding modelBoro . In particular we explore the band touching problem at high symmetry points. When we assume an identical transfer energy and an on-site energy , massless Dirac fermions emerge at two-band touching points ( and points), and different types of fermions emerge at three-band touching points (, , and points). In particular, fermions at the and points constitute pseudospin triplets separately. Then we construct an effective two-band theory or three-band theory in the vicinity of each touching point. Next, we consider the models together with realistic parameters for and . We consider two models with and without the inversion symmetry by an appropriate choice of the on-site energies. We find anisotropic massive Dirac fermions with the use of inversion nonsymmetric parameters. The degeneracy at the three-band touching points are slightly resolved both for the inversion symmetric and nonsymmetric models. Finally we study the edge states of borophene nanoribbons.
This paper is composed as follows. In Sec. II we review the basic properties of the lattice structure, the Brillouin zone and the symmetry for borophene. In Sec. III, we start with a five-band model comprised of the orbitals of Boron. We compare three types of models. One is a homogeneous model, where we take an identical transfer energy and an identical on-site energy. The second is the inversion symmetric model, where the transfer and on-site energies are chosen so as to respect the inversion symmetry. The third is the inversion nonsymmetric model, where on-site energies breaks the inversion symmetry. We show that Dirac fermions are gapless (gapped) when the inversion symmetry is present (absent). In Sec.IV, we derive an effective Dirac theory for general parameters and confirm the above results. In Sec.V, we derive effective theories of fermions at three-band touching points. It is shown that the set of fermions at the or point is unitary equivalent to the triplet of the pseudospin (). In Se.VI, we study edge states of borophene nanoribbons, where five different types of edges are introduced corresponding to the unit cell number.
II lattice
The lattice structure of borophene is illustrated in Fig.1(b). The unit cell contains five atoms as in Fig.1(a). The "a" and "e" atoms have four bonds, the "b" and "d" atoms have five bonds, and the "c" atoms have six bonds, leading to different on-site potentials.
The Brillouin zone is a rectangular given by and , as shown in Fig.2(a). However, it is convenient to use a shifted Brillouin zone and . The area of the Brillouin zone of borophene is one half of that of the honeycomb lattice. This is understood as follows. The lattice without the "c" atoms is identical to the honeycomb lattice, where the unit cell contains four atoms. On the other hand, the honeycomb lattice has only two atoms in the unit cell. Namely, the Brillouin zone of the borophene must be one half of that of the honeycomb lattice.
The symmetries of the lattice are the inversion symmetry , the two mirror symmetries with respect to the and axes, and . There is a relation . On the other hand, the rotation symmetry is absent in the lattice, which exists in the honeycomb lattice.
III Five-band model
A recent first-principles calculation demonstrates that the system is well described only in terms of the orbitals of the boron atomsBoro , implying that the tight-binding model is five dimensional,
[TABLE]
with
[TABLE]
The parameters obtained by fitting first-principles calculation results are summarized asBoro ,
[TABLE]
showing that the transfer energies are symmetric along the axis, and
[TABLE]
The lattice constant is Å. A characteristic feature is that the inversion symmetry of the lattice structure is broken by this set of on-site energies. We refer to this tight-binding model as the inversion nonsymmetric model.
We first consider the modelBoro by setting all transfer energy equal (eV) and all on-site energy zero (), which we refer to as the homogeneous model. We also investigate the inversion symmetric model, which is defined by the following set of the one-site energies instead of (4),
[TABLE]
where the magnitude of the on-site energy reflects the number of adjacent atoms in each sites. We show the band structures of the homogeneous model and the inversion nonsymmetric model in Fig.3(a1) and Fig.3(b1), respectively. We also show their project band structures along the axis in Fig.3(a2) and Fig.3(b2). Those for the inversion symmetric model are quite similar to these.
We start with the investigation of the homogeneous model: See Fig.3(a1). We find Dirac fermions at the two points , where the energy is explicitly obtained as
[TABLE]
with the use of a unitary transformation . (These two points are customarily called the and points.) It is interesting that there are different types of three-band touching points. Their positions in the Brillouin zone are shown in Fig.2(b). One is at the point , where the energy is given by
[TABLE]
The second one is at the point , where the energy is given by
[TABLE]
The third one is at the points , where the energy is given by
[TABLE]
We show the detailed band structures of Dirac fermions and triple-point fermions in Fig.4(a1),(b1),(c1) and (d1).
We may investigate both the inversion symmetric and nonsymmetric models in a similar way. Their overall band structures are very similar to that of the homogeneous model, as shown in Fig.3(b1) for the inversion nonsymmetric model. However, there arise differences with respect to the degeneracy at the band touching points. First, the Dirac fermions remains gapless in the inversion symmetric model but gets gapped in the inversion nonsymmetric model. On the other hand, three-band touching points are slightly resolved both in the inversion symmetric and nonsymmetric models. We show the detailed band structure at these points in Fig.4.
IV Dirac fermions
Since the dimension of the matrix in the tight-binding model (1) is five, it is impossible to diagonalize it analytically. It is highly desirable to construct such models with lower dimensions that we can analyze analytically. We construct an effective two-band Hamiltonian for Dirac fermions in this section and effective three-band Hamiltonians for fermions at three-band touching points in the next section.
It is reportedBoro that the amplitude of the zero-energy wave function at the and points is exactly zero at the "c" sites for the homogeneous model. Then, it is reasonable to neglect the "c" atoms in the Hamiltonian (1), and we obtain the following four-band Hamiltonian to describe the physics in the vicinity of the and points,
[TABLE]
The energy is analytically obtained as
[TABLE]
Indeed, as we show in Fig.5(a), it well reproduces the original band structure of the Dirac fermions both at the and points.
The corresponding lattice is a honeycomb lattice shown in Fig.1(c). The unit cell contains four atoms comprised of the "a", "b", "d" and "e" atoms. On the other hand, the unit cell of the honeycomb lattice contains two atoms such as "b" and "d". Hence the above four-band model can be further reduced to the two-band model.
We are able to construct actually the two-band model by way of in the vicinity of the Dirac points, where is the projection operator from the Hamiltonian to the Hamiltonian containing the two bands with the zero eigen-energy. The low-energy effective Hamiltonian is given by
[TABLE]
with
[TABLE]
It is identical to the Hamiltonian of the anisotropic honeycomb lattice with on-site potentials,Phos which corresponds to the lattice without the "c" atoms. This correspondence is due to the fact that the amplitude of the wave function at the "c" sites is zero at the zero energy.
The gap closes at
[TABLE]
when . Especially, the gap closes in the presence of the inversion symmetry since and . The gap closing point shifts from the original point when . The gap opens when with the gap . We estimate the gap is eV by using the parameters (4) in the inversion nonsymmetric model.
We show the band structure along the axis of the two-band model as well as the five-band model in Fig.5(b). The Dirac fermions at the point is well reproduced, while the Dirac fermions at the point disappear. This is due to the fact that the Brillouin zone is enlarged twice since the unit cell becomes half compared with that of the four-band model, as shown in Fig.2. It is necessary to construct another two-band model by expanding at the point, which is done precisely in the similar way.
In the vicinity of the point, the two-band Hamiltonian is expanded as
[TABLE]
which describe a Dirac cone. The dispersion is isotropic only for the homogeneous model.
In passing, it is intriguing to see that the four-band theory describes precisely two bands of the and points though it is constructed soley for the and points in Fig.5.
V Fermions at three-band touching points
We next construct three-band effective models for fermions at three-band touching points. We construct effective models as in the case of the Dirac fermions with the use of the unitary transformation and the projection to the low-energy bands.
X point: The effective Hamiltonian valid in the vicinity of the point is given by
[TABLE]
where
[TABLE]
in the case of the homogeneous model. See Appendix for general parameters.
We show the band structure in Fig.6(a1). The vicinity of the three-band touching point is well reproduced by this model. Furthermore, comparing the band structure of the three-band model along the line with that of the original five-band model , the three-band model is found to reproduce perfectly the two bands given by all over the region: See 6(a2).
We wonder why there is no partner for the point as in the case of the and points. We study this problem for the homogeneous model. The three-band Hamiltonian is expanded in the vicinity of the point as
[TABLE]
with . The corresponding wave functions are
[TABLE]
The Berry phase is zero for each band,
[TABLE]
Since the band carries no topological charge, the point can exist by itself. Furthermore, it indicates that the three-band touching point is not topologically protected.
The energy eigenstate of the Hamiltonian (18) is given by . Hence it is unitary equivalent to the following Hamiltonian,
[TABLE]
where is the pseudospin operator obeying , etc., whose magnitude is . Namely, the three bands are members of a pseudospin triplet with . On the other hand, for realistic and , the three-band touching point is resolved.
M point: In the vicinity of the point, we obtain the following effective three-band model,
[TABLE]
where
[TABLE]
in the case of the homogeneous model. See Appendix for general parameters.
We show the band structure in Fig.6(b1). In Fig.6(b2), comparing the band structure of the three-band model along the line with that of the five-band model , we find that the three-band model perfectly reproduces the two-bands given by all over the region. On the other hand, the middle band becomes a perfect flat band in the three-band model , while it is dispersive in the five-band model .
We expand the three-band Hamiltonian in the vicinity of the point as
[TABLE]
with . The energy is obtained as . Hence it is unitary equivalent to the following Hamiltonian,
[TABLE]
The three bands are members of a pseudospin triplet with . The Berry phase is zero for each band. Consequently, the point is not accompanied by a partner. These properties are quite similar to those of the point.
point*:* In the vicinity of the point, we obtain the following effective three-band model,
[TABLE]
with
[TABLE]
We show the band structure in Fig.6(c1). In Fig.6(c2), we compare the band structure of the three-band model along the line with that of the original five-band model , and find that the three band model perfectly reproduces the two bands given by all over the region.
In the vicinity of the point, the Hamiltonian is expanded as
[TABLE]
with
[TABLE]
and .
VI Borophene nanoribbons
When a nanoribbons is along the zigzag direction there are zigzag and beard edges in the case of the honeycomb system. This is because there are two atoms in the unit cell. There are five types of borophene nanoribbons with zigzag edges corresponding to the fact that the unit cell contains five atoms. For example, the edge terminated by "a","b", "c" and "d" atoms forms a zigzag edge, while that terminated by "e" atoms forms a beard edge. Thus there are different nanoribbons.
We show the band structure of typical nanoribbons in Fig.7(a)(e). In Fig.7(f), we show the bulk band structure projected to the axes for the sake of comparison. The band structure of nanoribbons are almost identical to those of the projected bulk band structure except for the edge states, which are marked by magenta curves. The edge states emerge in the region connecting between the Dirac point and the triple point. Among them, there emerge almost flat bands at the zero energy in Fig.7(e), which corresponds to the beard edge states. If the two terminations are different, the edge states are the sum of the two terminations.
VII Discussion
It is interesting that Dirac fermions or triplet fermions emerge in the structure of borophene at the high symmetry points. There is a distinctive difference between them. On one hand, Dirac fermions emerge always in a pair: They emerge at the and points just as in graphene. The reason is the Nielsen-Ninomiya theorem. Namely, the gapless Dirac fermion has Berry phase while the gapped Dirac fermion has Chern number. They appear in a pair so that the total topological number must be zero.
One the other hand, this is not the case for triplet fermions. There are no partners for the and points. Indeed, calculating the Berry phases of the bands at and , we find them to be zero.
The lattice structure of borophene has the inversion symmetry, where massless Dirac fermions are expected. However, the inversion symmetry is broken in the Hamiltonian together with the parameters (3) presented in Ref.Boro , where Dirac fermions are gapped. The ARPES experiment shows that the gap is absent within the experimental resolution.
We note that there is a metallic band at the zero-energy in the five-band model, while it is absent in the effective lower-band theories. It is because that they are valid in the vicinity of the high symmetry points. We should use the five-band model when we calculate the conductivity and others.
The author is very much grateful to N. Nagaosa for many helpful discussions on the subject. He thanks the support by the Grants-in-Aid for Scientific Research from MEXT KAKENHI (Grant Nos.JP25400317 and JP15H05854). This work is also supported by JST, CREST (Grant No. JPMJCR16F1).
Appendix: Three-band theories with general parameters
In this appendix we present the matrix elements of the effective three-band theories (16) and (5) with general parameters and .
X point:
[TABLE]
M point:
[TABLE]
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1(1) A.H. Castro Neto, F. Guinea, N.M.R. Peres, K.S. Novoselov, and A.K. Geim, Rev. Mod. Phys. 81 , 109 (2009).
- 2(2) C.-C. Liu, H. Jiang, and Y. Yao, Phys. Rev. B 84 , 195430 (2011).
- 3(3) M. Ezawa, Phys. Rev. Lett 109 , 055502 (2012).
- 4(4) L. Tao, et.al., Nature Nanotechnology 10, 227 (2015)
- 5(5) M. E. Davila, L. Xian, S. Cahangirov, A. Rubio, G. Le Lay, New Journal of Physics 16 (9), 095002
- 6(6) L. Li, et. al., Adv. Mater. 26, 4820 (2014)
- 7(7) M. Derivaz, et.al., Nano Lett. 15, 2510 (2015)
- 8(8) F. Zhu, et.al., Nat. Mater. 14, 1020 (2015)