TDDFT+U: Hubbard corrected approximate density-functional theory in the excited-state regime
Okan K. Orhan
David D. O’Regan
School of Physics, Trinity College Dublin, Dublin 2, Ireland
Abstract
We develop a generalization of the Kohn-Sham density functional theory (KS-DFT) + Hubbard U (DFT+U)
method to the excited-state regime. This has the form of Hubbard U corrected linear-response time-dependent DFT, or ‘TDDFT+U’.
Combined with calculated linear-response Hubbard U parameters, it may provide a computationally light, first-principles method for the simulation of tightly-bound excitons on transition-metal ions.
Our presented implementation combines linear-scaling DFT+U and linear-scaling TDDFT, but the approach
is broadly applicable.
In detailed benchmark tests on two Ni-centred diamagnetic coordination complexes with variable U values, it is shown that the Hubbard U correction to an approximate
adiabatic semi-local
exchange-correlation interaction kernel lowers the excitation energies of transitions exclusively within the targeted localised subspace, by increasing the exciton binding of the
corresponding electron-hole pairs.
This partially counteracts the Hubbard U correction to the exchange-correlation potential in KS-DFT,
which increases excitation energies into, out of, and within the targeted localised subspace by modifying the underlying KS-DFT eigenspectrum.
This compensating effect is most pronounced for optically dark transitions between localized orbitals
of the same angular momentum, for which experimental observation may be challenging and theoretical approaches are at their most necessary.
Interestingly, we find that first-principles TDDFT+U
seems to offer a remarkably good
agreement with experiment for a
perfectly closed-shell complex on which approximate
TDDFT under-performs, but only when TDDFT+U
is applied to the DFT eigenspectrum
and not to the DFT+U one.
In tests on an open-shell, non-centrosymmetric, high-spin cobalt coordination complex, we find that
first-principles TDDFT+U again compensates
for the DFT+U blue-shift
in 3d→3d transitions,
but that using the DFT eigenspectrum is not viable
due to the emergence of a singlet instability.
Overall, our results point to shortcomings in the
contemporary DFT+U corrective potential,
either in its functional form, or when
applied to transition-metal orbitals but not to ligand
ones, or both.
I Introduction
Density-functional theory (DFT) Hohenberg and Kohn (1964); Kohn and Sham (1965)
provides a computationally tractable means by which
to investigate the quantum-mechanically derived properties of
molecules and materials.
TDDFT Runge and Gross (1984) is its elegant extension
to the dynamical, excited-state regime.
TDDFT is now widely used to investigate
the excitation spectra of extended solids and molecules alike Petersilka et al. (1996); Bauernschmitt et al. (1997); Stratmann et al. (1998), due to its relatively low computational
cost relative to wave-function and Green’s function based approaches.
While DFT and TDDFT are both exact in principle,
their accuracies in practice are limited
by the approximations currently available
for the exchange-correlation (xc) contribution to the total-energy
functional Exc and its derived
interaction kernel (by second functional derivatives), fxc.
Common xc-functionals include local functionals such
as the local density approximation Kohn and Sham (1965),
semi-local functionals such as generalized gradient approximations Perdew et al. (1996), and semi-empirical functionals
such as hybrids Kim and Jordan (1994); Adamo and Barone (1999); Heyd et al. (2003).
In practice, an adiabatic, i.e., time-averaged interaction approximation
is made to construct the xc-kernels
of contemporary applied TDDFT.
The latter is often also restricted, for
expediency, to the
linear-response regime appropriate only to low-energy, low-oscillator-strength excitations.
I.1 Self-interaction error in approximate DFT
and its correction by Hubbard U based methods
Perhaps the most transparent systematic error exhibited by approximate functionals is the single-particle self-interaction error (SIE) Cohen et al. (2008a),
i.e. the tendency of electrons to effectively self-repel,
and has been demonstrated clearly in single-electron systems such as the molecule H2+ Merkle et al. (1992); Savin (1996); Perdew and Levy (1997); Zhang and Yang (1998).
This error becomes more complicated in the many-body case and hence, by necessity, there has emerged the more general concept of many-body self-interaction error Perdew and Zunger (1981), also known as delocalisation error Mori-Sánchez et al. (2006a, b); Vydrov et al. (2006); Ruzsinszky et al. (2006, 2007); Cohen et al. (2012), which has been developed to understand the
collective spurious self-interaction of approximated electron
densities.
In a system with a continuously variable occupation number,
many-body SIE may be defined
as the deviation from piecewise linearity of the approximate DFT total-energy with respect to the total electron count Perdew et al. (1982).
The SIE is most problematic for systems
comprising spatially localized, partially filled frontier
orbitals including those of 1s and 2p but
more canonically 3d and 4f character,
where the qualitative failure of local and semi-local functionals has been thoroughly
analysed Austin and Mott (1970); Terakura et al. (1984); Anisimov et al. (1991); Cococcioni and de Gironcoli (2005).
First-row transition metals systems thus
can often
benefit from corrective measures that augment conventional
closed-form density functionals.
An approach that is very widely used at present
is the computationally expedient DFT+U,
which has been successfully applied to both extended solids Austin and Mott (1970); Anisimov et al. (1991, 1993); Solovyev et al. (1994); Liechtenstein et al. (1995); Anisimov et al. (1997); Pickett et al. (1998); Himmetoglu et al. (2011) and molecular systems Scherlis et al. (2007); O’Regan et al. (2010); Kulik and Marzari (2010, 2011); Cole et al. (2012); Himmetoglu et al. (2014) alike.
DFT+U attains the status of a first-principles
method through the direct
calculation of the requisite Hubbard U parameters,
and for which a number of methods
have been proposed Pickett et al. (1998); Cococcioni and de Gironcoli (2005); Kulik et al. (2006); Aryasetiawan et al. (2006); Şaşıoglu et al. (2011); Himmetoglu et al. (2014).
We refer the reader to Ref. 43 for a recent detailed
analysis of Hubbard U and Hund’s J (the analogous quantity for
quantifying erroneous energy-magnetization curvature in approximate DFT)
calculation in the case of open-shell systems.
DFT+U is compatible with linear-scaling
methods Han et al. (2006); O’Regan et al. (2012)
intended for spatially complex systems, as well as with high-throughput materials
discovery approaches Curtarolo et al. (2013); Agapito et al. (2015).
Beginning with Ref. Kulik et al., 2006,
and continued in
Refs. Kulik and Marzari, 2010; Himmetoglu et al., 2011; Kulik and Marzari, 2011; Zhao et al., 2016,
the concept of DFT+U as a corrective method
for SIE has been extensively developed,
with the Hubbard U parameters playing the role of localized
error quantifiers of SIE for the approximate
functional applied to the specific system at hand Cococcioni and de Gironcoli (2005).
We invoke this interpretation in what follows.
I.2 Self-interaction error in the excited-state regime
For the integer-occupancy systems routinely simulated,
the generalized Koopman’s condition Dabo et al. (2010)
gives a unified, practicable expression for the SIE-free
condition, the non-compliance with which is, in most cases,
responsible for the underestimated insulating
gaps Cohen et al. (2008b, 2012)
emblematic of practical DFT.
When this cannot obviously be enforced, however, such as in neutral excited states,
it will be helpful to decompose SIE into two contributions.
The first is an overestimation of the net self-repulsion
of the electron density due to the spurious self-interaction of
individual electron densities, particularly so for localized
atomic orbitals, which gives rise to a positive
energy-occupancy curvature, over-delocalised of densities,
and inaccurate ground-state total energies.
The second is the lack of any distinction
between the density due to electrons already existing
in a system and that due to any newly removed or added electrons,
which results in the spurious absence of derivative
discontinuities in the energy-occupancy curve
and, consequently, the shallowing of electron removal and addition levels
and the underestimation of insulating gaps.
Adiabatic linear-response TDDFT inherits
both components of SIE from the underlying approximate
DFT functional.
In this work, we will focus on the former
component while treating the latter
only at the level available
within first-principles DFT+U.
Technically, we use DFT+U in its
simplified rotationally-invariant formalism
(which does
not introduce a derivative discontinuity
but emulates the effects of one in the
Kohn-Sham Kohn and Sham (1965) eigenspectrum),
with first-principles
linear-response Hubbard U and
Hund’s J parameters.
The effect of SIE on electron dynamics
and neutral electronic excitations, such as those
routinely studied using TDDFT, has
slowly attracted increasing investigation
in recent years Ullrich et al. (1998); Messud et al. (2008); Hofmann et al. (2012); Hofmann and K mmel (2012).
It is a matter of central importance, for
example, in the first-principles simulation of out-of-equilibrium nanoscale
functionalities such as dynamical
Coulomb blockade Hofmann and Kümmel (2012); Hodgson et al. (2013),
and in the first-principles spectroscopy of systems comprising
transition-metal ions Autschbach et al. (2003); Chiodo et al. (2011); Pastore et al. (2013); Berardo et al. (2014a, b); Niehaus et al. (2015).
In the realm of non-atomistic calculations, the TDDFT
solution of Hubbard type models have also attracted
attention Verdozzi (2008); Farzanehpour and Tokatly (2014); Fuks and Maitra (2014); Magyar (2009),
and TDDFT has also been combined
with dynamical
mean-field theory Karlsson et al. (2011); Acharya et al. (2016).
I.3 Motivation:
Hubbard U correction in the excited-state regime of TDDFT
Somewhat surprisingly, perhaps, given its relatively
moderate computational cost and conceptual simplicity,
the error correction of approximate
TDDFT by means of DFT+U, in the guise of
adiabatic TDDFT+U, has received relatively little attention
to date.
TDDFT+U is readily compatible with linear-scaling DFT, as demonstrated in the present work though the combination of linear-scaling DFT+U O’Regan et al. (2012); Han et al. (2006) and linear-scaling TDDFT Zuehlsdorff et al. (2013, 2015, 2016), as well as with high-throughput materials screening techniques, where DFT+U is commonplace Curtarolo et al. (2013).
Within its range of applicability,
TDDFT+U could potentially offer substantial
efficiency advantages over more involved
methods for calculating neutral excitations in complex transition-metal
molecules and solids.
These include hybrid
TDDFT Hay (2002); Rosa et al. (2004)
and Green’s function based methods such as GW +
Bethe-Salpeter Körbel et al. (2014).
Recently, the optimally-tuned, range-separated hybrid functionals Savin (1995); Leininger et al. (1997) within TDDFT have met with promising success in the prediction of optical excitations, particularly in the lowest excitations in organic molecules and third-row transition-metal coordination complexes Kronik et al. (2012); Jacquemin et al. (2014); Bokareva et al. (2015).
This latter approach has been not applied to any first-row transition-metal molecules yet, to our knowledge.
The role of DFT+U in calculated excitation energies,
particularly the explicit contribution from the Hubbard term,
has been explored in Ref. Himmetoglu et al., 2012.
The first reported TDDFT+U implementation was that of Ref. Qian et al., 2009, combining real-time propagation and a plane-wave basis, followed by Ref. Lee et al., 2010, which detailed the results of a linear-response implementation applied to bulk NiO.
In that system, TDDFT+U was shown to be capable of reproducing the experimentally observed, tightly-bound Frenkel excitons,
but not their multiplet structure.
These are relatively exotic spectroscopic features that neither the adiabatic LDA, nor the
random phase approximation built from LDA+U,
succeeded in recovering to any extent.
Recently, in Ref. Shin et al., 2016, a real-time
plane-wave TDDFT+U implementation has been coupled
with Ehrenfest molecular dynamics to simulate both long and
short-ranged dynamical charge-transfer between alkali
atom impurities and conjugated carbon systems.
This work revealed
the tendency for an increasing Hubbard U
to promote the availability of multiple low-energy
states in such systems, as well as to increase
in energy and broaden the impurity-bath
charge-transfer resonances.
To date, however, information has been lacking on how
the Hubbard U correction affects the typical
products of practical TDDFT calculations in
simple transition-metal systems, namely
the low-energy excitation spectra and dipole-dipole absorption
spectra, for better or worse with respect to experiment.
Indeed, the precise effects of TDDFT+U
have yet to be systematically studied, and
its resulting range of applicability has yet to be
mapped out in any sense.
It is
this knowledge gap
that we seek to
begin to fill
with the present exploratory study.
I.4 Outline of the paper: systematic
decomposition of the effects of Hubbard U correction in Kohn-Sham
DFT and linear-response TDDFT
We seek to
systematically investigate the role of DFT+U
as it separately alters the Kohn-Sham eigenspectrum
underlying a linear-response TDDFT calculation, and the
TDDFT interaction kernel itself.
For this,
following its detailed introduction via an
illustrative four-level
toy model in Section II,
we uncover the effects of full
TDDFT+U, in Section III,
on two representative diamagnetic
nickel complexes
(one perfectly closed-shell, one less so),
which were chosen for study due to
their relatively simple coordination chemistry.
Since their Ni 3d sub-shells are close to being
fully filled, nominally,
the dominant errors in the description of these molecules
using an approximate semi-local xc-functional
(in this work always Purdew-Burke-Ernzerhof, PBE Perdew et al. (1996))
and xc-kernel (adiabatic PBE) may be
ascribed primarily to SIE
(electron delocalization) rather than static
(multi-reference) correlation
error Cohen et al. (2008a, b).
For these systems, in Section IV,
we show that
first-principles Hubbard U
correction at the TDDFT level alone, leaving the
underlying Kohn-Sham eigenspectrum at its DFT level,
offers a far better agreement with available experimental
and quantum-chemical data, when compared to either uncorrected DFT & TDDFT
or consistent DFT+U & TDDFT+U.
Performing Hubbard U correction at the DFT level alone
meanwhile,
leaving the TDDFT kernel uncorrected,
leads to very unreasonable results indeed.
We will discuss some implications and possible solutions
to this intriguing asymmetry in Section VI.
We will turn first, however, in Section V, to the technically challenging case of an
open-shell system, a non-centrosymmetric,
high-spin cobalt coordination complex.
Here, we will again find that a first-principles
DFT+U correction applied only
to the Kohn-Sham eigenspectrum
drastically degrades the agreement between
the singlet excitation and the dipole-dipole absorption spectra
and, respectively, high-level
quantum-chemical and experimental data.
The agreement is recovered to some degree when
TDDFT+U is also used, but a number of important
spectral features remain poorly described.
In this case, we will show that the application of
first-principles TDDFT+U upon the
DFT Kohn-Sham eigenspectrum is not a viable
work-around, as the implied
inconsistency leads to
the emergence of a singlet instability.
II Hubbard correction of the exchange-correlation kernel: theory and numerical illustration
Let us now introduce the anatomy of the
Hubbard U correction to approximate TDDFT.
Concerning ourselves only with low-energy
single-particle excitations,
we will restrict ourselves to the linear-response regime.
Here, the spin-unpolarized TDDFT problem may
be expressed in the occupied-unoccupied Kohn-Sham
eigenvector product space via Casida’s equation Casida (1996, 2009), which is an
eigen-equation for the vertical
excitation frequencies ω, given
in its canonical notation by
[TABLE]
The
Hamiltonian
matrix elements
Acv,c′v′=δvv′δcc′ωc′v′+Kcv,c′v′ and Bcv,c′v′=Kcv,v′c′ correspond to
excitation-excitation pairs and
excitation-relaxation pairs, respectively.
The neglect of coupling between these
processes, that is the approximation B=0, is known as the Tamm-Dancoff
approximation (TDA).
The ground-state Kohn-Sham
eigenvalues ϵv are those of occupied valence states,
while the ϵc are those of unoccupied conduction states.
The coupling matrix K incorporates all interactions
between particle-hole pairs, which is to say all effects
beyond the many-body random-phase approximation
(Fermi’s Golden Rule, or FGR).
It is given, within the valence-conduction (cv) product
representation of the interaction kernel f^, by
[TABLE]
where the
ψ are Kohn-Sham eigenvectors.
The kernel ordinarily comprises Hartree and xc
terms only, denoted by f^H
and f^xc,
but if a DFT+U derived correction term f^U is added,
the resulting TDDFT+U interaction kernel is given by
f^=f^U+2(f^H+f^xc).
The underlying Kohn-Sham eigensystem is also changed, typically.
The factor of 2 here is conventional,
and it represents the sum of
identical (in the unpolarized case)
like and unlike-spin Hartree and xc interactions
acting on a given excitation.
This factor of 2 does not, however, pre-multiply
f^U, since DFT+U ordinarily acts explicitly
only on like-spin Kohn-Sham states.
The rotationally-invariant DFT+U energy
functional Anisimov et al. (1991, 1993); Solovyev et al. (1994); Liechtenstein et al. (1995); Anisimov et al. (1997)
used in this work falls into this category,
being given, for a SIE-affected subspace, by
[TABLE]
where Ueff=U−J is the effective like-spin correction parameter expressed in terms of the Hubbard U and the Hund’s J parameter. The index σ is for spin, and the subspace occupancy matrix
nmm′σ=∑v⟨φm∣ψvσ⟩⟨ψvσ∣φm′⟩
is typically defined in terms of localized
orbitals (in our calculations, orthonormal
atomic nickel or cobalt 3d orbitals solved
in a norm-conserving pseudopotential), φm.
The Hubbard U kernel
is the second functional derivative Runge and Gross (1984)
of the DFT+U energy EU with respect to the
density matrix, and we find,
denoting the density-matrix for spin σ by
by ρσ(r,r′), that
[TABLE]
The resulting Hubbard U contribution to
K may be written, using implicit
summation of paired indices, as
[TABLE]
whereafter we will use U rather Ueff for simplicity, except where discussing our actual calculated
Ueff.
The resulting ‘direct’ term, in what can be seen as an effective
exciton self-interaction
correction, is given by
[TABLE]
The form of KU hints at the behaviour expected
of the TDDFT+U excitation spectrum as U is varied.
For U>0 eV, the interaction correction
due to one (cv) pair and acting upon another
is a sum over (typically) attractive direct Hartree and
exchange terms.
Relative to the situation that holds in hybrid-exchange TDDFT, however,
the exchange terms are expected to be more significant relative to
direct Hartree ones,
since in TDDFT+U the same constant U pre-multiplies both
term types.
It is instructive to examine the special case in which
the projecting orbitals φm are identical to a subset of
the underlying Kohn-Sham states ψ.
There, the Hubbard U contributions to B and A
reduce considerably to
[TABLE]
leaving a fully diagonal contribution to the Casida Hamiltonian.
If these Kohn-Sham states are also well separated from all others
energetically, the effect of the Hubbard U on the underlying
eigenstate differences ϵc−ϵv will simply be an
increase by U, whereupon the effects of DFT+U
and TDDFT+U fully cancel for excitations coupling states
within the target subspace.
This picture is complicated
by Kohn-Sham state hybridization, self-consistency, and the spillage of
the localized orbitals, in practice.
Nonetheless, the TDDFT+U correction may be expected
to increase the mixing of transitions between states
that overlap strongly with the selected subspace,
and to increase their exciton binding energy by
compensating for the underlying DFT+U eigenvalue correction.
However, the matrix elements of KU are
quadratic in overlap integrals of the form
⟨ψc∣φ⟩⟨φ∣ψc⟩,
whereas the underlying Hubbard U correction to the Kohn-Sham
potential comprises terms that are only linear in such integrals.
Thus, we cannot generally expect the cancellation of
the U correction to the ground and excited-state
systems to be precise in practical calculations.
II.1 Illustration of the effect of U correction in TDDFT using a four-level toy model
For further insight, the effects of TDDFT+U in conjunction with DFT+U can be illustrated by means of a toy model in conjunction with the TDA and full Casida equation.
Let us consider four independent-particle (KS-like) states, of which two occupied and two unoccupied states are labelled with {v,v′} and {c,c′}, respectively, with some arbitrary eigenenergies as illustrated in Fig. 1.
The pair {c,v} of states shown in dashed-red are targeted with a correction inspired by DFT+U and TDDFT+U.
The block matrices A and B in the Casida equation become 4×4 matrices with elements
given by
[TABLE]
where j and j′ run over {c,c′}, while i and i′ run over {v,v′}.
The Hubbard parameter UDFT imitates the effect of DFT+U by pushing the targeted states away from the Fermi level via the term UDFT(δj′c+δi′v)/2, whereas the Hubbard parameter UTDDFT includes the effect of TDDFT+U via the term −UTDDFTδj′i′,cv.
By making these two Hubbard parameters UDFT and UTDDFT independent, the individual effects of the Hubbard corrections at the DFT and TDDFT levels can be observed by setting one of them to zero at a time.
The Hartree+xc coupling matrix elements are assigned for illustration here
to the arbitrary values
[TABLE]
and the symmetric choice made here
is a deliberate attempt to simplify the contributions due to fHxc.
The Casida equation, both in its full form and within the TDA, was solved using an eigenvalue solver over a range of
UDFT and UTDDFT values.
Additionally, FGR excitations energies are included and calculated as
[TABLE]
In Fig. 2, the principal effects of a positive UDFT
(simulating DFT+U) and UTDDFT (simulating TDDFT+U)
in our toy model
are demonstrated, via the amplitudes of normalised
electronic excitation spectra
(EES) calculated using Eq. (19).
A life-time broadening of Γ=0.1 eV
was used here, together with a high-resolution grid of Hubbard U parameters taken in 0.05 eV steps.
Starting from the energy levels shown in Fig. 1, a positive value of U=UDFT pushes the targeted (red-dashed in Fig. 1) states (v,c) away from the Fermi level, each by with U/2, while the bystander states remain intact.
Consequently, in Fig. 2a, the excitation from v to c (v→c) increases simply by U, while the energies of v′→c and v→c′ increase by U/2, emulating the effects of DFT+U.
The remaining excitation v′→c′ is not affected due to lack of interaction between exciton pairs within FGR.
Comparing next Figs. 2b, 2c, and 2d against the FGR results of Fig. 2a, taken each at U=0 eV, a global shift
by TDDFT of ∼3−4 eV on the excitation energies can be seen, as
well as the avoided crossing of excitation energies for U>0 eV.
This is due to the interactions between exciton pairs,
emulating TDDFT, that are introduced by the coupling matrix Kji,j′i′Hxc in Eq. (II.1).
The global nature of the shift is due to the invariance of the coupling matrix with respect to the swapping of orbital indices.
In Figs. 2c and 2e,
the UTDDFT term (emulating TDDFT+U) exclusively affects the excitation v→c by pushing it down
(linearly in the TDA case)
from ≈8 eV for increasing U=UTDDFT values.
For U=4 eV (U=8 eV for TDA), the excitation v→c becomes purely imaginary
(negative in the TDA case),
meaning that the model becomes unphysical.
In Fig. 2d, the combined emulated
effects of DFT+U and TDDFT+U, when UDFT=U=UTDDFT, are seen in the form of a total cancellation of the effect of DFT+U on the excitation v→c by TDDFT+U.
The remaining three excitations are affected by DFT+U
as before, while the effect of TDDFT+U (comparing
Figs. 2b and 2d)
is relatively minor and mostly due to avoided crossing.
Comparing Fig. 2d with its TDA counterpart
Fig. 2f, the excitations within this model show a similar
qualitative behaviour
irrespective of whether the
TDA is invoked.
The TDA approximately shifts the excitations up in energy by
∼1 eV throughout the frequency range.
II.2 Implementation of the TDDFT+U kernel
within linear-scaling linear-response TDDFT
We have implemented the TDDFT+U kernel of
Eq. 10 in the ONETEP
package Skylaris et al. (2005); Haynes et al. (2006); O’Regan et al. (2012).
This direct-minimization DFT
code maintains a linear-scaling increase in computational
expense with respect to system size, while maintaining
an accuracy which is effectively
equivalent to that of a plane-wave code.
It does this by expanding the Kohn-Sham density-matrix in terms
of a minimal set of spatially truncated
non-orthogonal generalized Wannier functions (NGWFs),
which are variationally optimized in situ Skylaris et al. (2002).
For calculations involving excited states,
the code is capable of variationally optimizing a
set of Wannier functions for the unoccupied conduction bands
as a post-processing step that follows conventional total-energy
minimization Ratcliff et al. (2011).
With this, and using the resulting joint basis of optimized
valence and conduction band Wannier functions,
we used the linear-scaling beyond-Tamm-Dancoff linear-response
TDDFT functionality
available in ONETEP
Zuehlsdorff et al. (2013, 2015, 2016), which again uses iterative minimization,
as the basis for our implementation.
The central element in our combination of linear-scaling TDDFT
and DFT+U O’Regan et al. (2012) is the
change in DFT+U potential associated with the
first-order change in Kohn-Sham density-matrix,
ρ(1)(r,r′;ω) at a each excitation energy ω,
which is given by the same expression
for both singlet and triplet excitations alike, specifically
[TABLE]
From this equation, it is clear that the occupancy dependence of the
DFT+U potential survives in TDDFT+U, insofar
as that, for U>0 eV,
a level within the target subspace that is depopulated
under excitation (typically a valence level close to the gap)
will be subject to a more repulsive DFT+U
potential, whereas a repopulated (e.g., conduction)
level will be subject to a more attractive
DFT+U potential.
TDDFT+U thus tends to promote such excitations by
increasing the exciton binding between the associated levels.
We emphasise that the interaction
in TDDFT+U remains entirely adiabatic
as it is presented here, since the kernel
f^U is constant, and so it addresses
only the time-average of the self-interaction error as it is
measured in the ground-state.
As a result, it lacks the ability to produce dynamical step
features in the potential that may result of occupancies
passing through integer values, which are dynamical
manifestations of the second aspect of self-interaction error
previously discussed.
However, TDDFT+U does provide a convenient framework
in which to explore non-adiabatic self-interaction
correction kernels f^U(ω),
either by means of an explicitly frequency-dependent Hubbard
U(ω).
III The Hubbard U dependence of
neutral excitation spectra
Two small closed-shell Ni-centred coordination complexes, namely the planar tetracyanonickelate anion Ni(CN)42-
and tetrahedral nickel tetracarbonyl Ni(CO)4 shown in Fig. 3, were chosen for study.
The Hubbard U dependence of molecular spectra, in
terms of both its individual effects on DFT+U and TDDFT+U,
and on their combination,
was investigated.
These systems provide a useful playground in which to investigate the effects of DFT+U and TDDFT+U, since they minimise any complex contributions from magnetic ordering and large ligand-field splittings, as both systems are closed-shell and centro-symmetric with strong ligands.
Furthermore, these systems have previously been studied experimentally Gray and Ballhausen (1963); Schreiner and Brown (1968); Lever et al. (1979); Kotzian et al. (1989) and using numerous first-principles methods Pierloot et al. (1996); van Gisbergen et al. (1999); Hummel et al. (2006); Shu et al. (2015).
This is not, however, to imply that these systems are
ideal candidates for treatment using DFT+U, let
alone TDDFT+U, since they are reasonably well
described by conventional approximate DFT.
Convention for visualising spectra
At this juncture we must introduce
our conventions for visualising two
essential molecular spectroscopies.
Electronic excitation spectra (EES) are constructed here by including both optically allowed and forbidden excitations with the same unit oscillator strength. They are calculated using the formula
[TABLE]
where ωji denotes the energy of a transition
from an occupied (i) to an unoccupied
(j) molecular electronic state, and
Γ is a Lorentzian broadening factor.
Electric dipole-dipole absorption spectra are
commonly
used to measure the optical response of molecules in the low-energy spectral range.
The contributions of the individual excitations are weighted by oscillator strengths
fj←i
related to the transition dipole moments.
The formula relevant to optical absorption is
[TABLE]
and this type of spectrum is
the one primarily used here for comparing with experimental observations.
EES and ABS were constructed using Eq. (19) and Eq. (20) with a Lorentzian broadening Γ=0.1 eV at integer values of the Hubbard U parameters, and interpolated to intermediate values in 0.01 eV steps.
Our EES are scaled by setting the global maximum of EES data across DFT & TDDFT, DFT+U & TDDFT, DFT & TDDFT+U, and DFT+U & TDDFT+U
to unity.
Similarly, our ABS are scaled by setting the global maximum of ABS data across all of these four combinations to unity.
Such separate scaling factors enable us to compare relative intensities within various methods as well as to maintain the comparability between EES and ABS within same method.
EES calculated within the FGR are scaled separately, using their own maxima.
III.1 The square-planar tetracyanonickelate anion: Ni(CN)42−
The square-planar Ni(CN)42− is a low-spin coordination complex, with a Ni center of nominal charge 2+.
(CN)- is a strong-field π-acceptor ligand that leads to ligand-splitting at 3d-levels of Ni, following dyz≈dxz<dxy<dz2<dx2−y2, where 3d8 electrons occupy the first four levels and the remaining 3dx2−y2 forms an dsp2-hybrid with the ligands in the square-planar symmetry Griffith (1964).
As a result, the low-lying excitations are expected to be predominantly of a mixed 3d→3d and metal-to-ligand 3d→π∗ character, as suggested by previous studies Hummel et al. (2006).
The energy alignment of 3d states is shown as a function of U in Fig. 4a.
For increasing U values, the occupied 3d states move to deeper energies.
The states close to the HOMO-LUMO gap (shown with red, dashed lines), which strongly contribute to low-lying excitations, fall to lower energetic states entirely at about U⪆7 eV.
Thus, low-lying excitations are pushed upwards and, ultimately, they combine with higher energy excitations of metal-to-ligand character, as seen in the EES
calculated using FGR in Fig. 4b.
Up to this point, the Hubbard U has been used only to modify the under-lying KS-DFT states via DFT+U.
In Fig. 5, a more complete and consistent picture is provided, by the EES for the first 50 singlet excitations calculated using various combinations of DFT+U and TDDFT+U.
In Fig. 5a, we see that an increasing U value in DFT+U reduces the 3d → 3d character of the excitations, and combines them with excitations from deeper states, similarly to the FGR case.
Beyond that, DFT+U is effective globally insofar as that it pushes other excitations to higher energies as well, by means of modifying the metal-to-ligand energy as seen in Fig. 4a.
On the contrary, in Fig. 5b,
we observe that TDDFT+U affects only the excitations of 3d→3d character,
while, as anticipated, the remaining excitations remain largely unaffected.
Furthermore, the affected excitations become non-physical for U⪆7 eV in DFT & TDDFT+U, similarly to what is observed in the four-level toy model.
This situation arises by virtue of exciton over-binding, where for
large values of U, the TDDFT+U contributions to coupling matrix elements Kcv,cvU in Eq. (II) over-compensate for the sums of energy differences ωcv and the Hartree+exchange-correlation contribution to coupling matrix elements, leading to
unphysical complex eigenvalues.
In Fig. 5c
we find that, when DFT+U and TDDFT+U are combined consistently, TDDFT+U primarily cancels the effects of DFT+U on 3d→3d type of excitations, which are in the ∼ 3.5 - 4.5 eV range.
This cancellation of DFT+U by TDDFT+U gives rise to an approximately quadratic net dependence on U within the full Casida equation,
as opposed to a rather linear net behaviour with U when the TDA is invoked.
We can clearly observe this when comparing Fig. 5c and TDA in Fig. 5d.
This, again, reflects what was previewed in our four-level toy model.
Overall, on one hand DFT+U is very efficient at modifying the ABS as it pushes low-lying optical transitions to higher energies, as seen in Fig. 6a, Fig. 6c and Fig. 6d.
On the other hand, TDDFT+U does not have any significant effect
at all on the ABS shown in Fig. 6b, as TDDFT+U acts solely on 3d→3d excitations, which are optically
perfectly dark in Ni(CN)42− here due to its idealized square-planar symmetry.
III.2 The tetrahedral nickel tetracarbonyl: Ni(CO)4
The tetrahedral Ni(CO)4 is another low spin coordination with a neutral Ni center,
but it is not perfectly isoelectronic with
Ni(CN)42−
as it has an uncomplicated, full 3d sub-shell.
The (CO)- ion is a strong-field π-acceptor ligand, which splits the 3d states of Ni into dz2≈dx2−y2<dxy≈dxz≈dyz due to the tetrahedral symmetry present.
The two-fold and the three-fold degenerate 3d splitting can be clearly distinguished by the differing response to DFT+U seen in Fig. 7a. In this systems, the low-lying singlet excitations are necessarily of a predominantly Ni 3d→π∗ character Kotzian et al. (1989); McKinlay et al. (2015).
In Fig. 7a, we observe that the two-fold degenerate dz2≈dx2−y2 states (red, dashed line) at −2 eV and the three-fold degenerate dxy≈dxz≈dyz states (red, dashed lines), at −3 eV for U= 0 eV, are pushed deeper with increasing U values within DFT+U.
In Fig. 7b, these immediate effects of DFT+U on the low-lying 3d→π∗ excitations,
at ∼4.0−5.5 eV for U=0 eV,
are reflected in up-shifts in the
FGR singlet EES with increasing U values.
Such shifts are larger for excitations from the dz2 and dx2−y2 states, as these are lowered more by DFT+U.
A complete picture of the behaviour of the first 50 excitations with
DFT+U and TDDFT+U is presented in Fig. 8.
The increasing U parameter in DFT+U affects excitation energies globally, by pushing them to higher energies.
In Fig. 8a, particularly, the excitations from the lower-lying 3d levels (dz2/dx2−y2→π∗), at ∼5−6 eV for U= 0 eV, climb most strongly and cross over with the excitations from the deeper states at around U≈ 4 eV, as was previewed in Fig. 7b.
A similar trend is also present with DFT+U as it is more effective on the excitations from the lower energetic 3d levels, as seen in Fig. 8b, where some cross over occurs with the lower-energy group of excitations.
The cancellation of DFT+U effects by TDDFT+U is more subtle in Ni(CO)4 for the relevant excitations compared to the situation in Ni(CN)42−, and this
(shown in Fig. 8c)
is as expected due to the weaker 3d→3d character of the transitions.
While TDDFT+U shifts the lowest group of excitations as well as splitting these excitations, it does not lead to the splitting-off of distinct tightly-bound excitons as observed in Ni(CN)42−.
As the dominant optically allowed transitions are almost purely of 3d→π∗ character, DFT+U naturally pushes bright excitations up in energy, as seen in Fig. 9a, whereas the effect of TDDFT+U on these excitations is quite subtle, which can be seen in Fig. 9b.
An important point to recall here is that, while DFT+U is effective in proportion to the 3d character of the KS manifold, TDDFT+U is proportional to the 3d character of product space of occupied 3d-unoccupied 3d subspaces.
IV First-principles spectra of two low-spin Nickel-centred complexes
The EES and ABS of our two closed-shell coordination complexes were generated using DFT+U and TDDFT+U with their respective first-principles Hubbard Ueff parameters,
following the detailed procedure described in the Appendices.
In particular, these spectra were obtained
by evaluating, or ‘slicing’, the interpolated data shown in the graphs presented in
Sec. III.1 and Sec. III.2, at the corresponding
first-principles Hubbard U parameters summarised in Table 5.
IV.1 Excitation energies and spectra of Ni(CN)42−
The EES and ABS of Ni(CN)42− are presented in Fig. 10 and Fig. 11 for the first-principles Ueff=U−J=6.901 eV, alongside experimental excitation spectra extracted from Ref. 90.
In Fig 11,
the experimental excitation peak positions are shown with vertical grey lines,
with heights indicating their relative absorbances with respect to that of
the experimental maximum absorbance at 4.66 eV, which is set to unity.
Excitation energies are listed in Table 1 along with the experimental
results Gray and Ballhausen (1963) and TDDFT results Hummel et al. (2006),
with optically bright excitations are highlighted with a bold font.
In particular, our
first-principles excitation energies were obtained from the peak positions of Fig. 10, with smaller peaks and shoulders removed, and the optically bright ones were assigned by matching to the peaks of Fig. 11.
The previous TDDFT calculations of Ref. 96 were performed using implicit solvation with a dielectric constant of 37.5,
whereas ours were performed under vacuum conditions.
Nonetheless, the former data provides an useful
benchmark for testing the numerical validity of our TDDFT+U code.
As seen Fig. 10, DFT+U is effective throughout the spectral range.
It shifts excitation features to higher energies, as seen by comparing DFT+U & TDDFT with DFT & TDDFT (PBE).
TDDFT+U, however, acts only in the low-energy range,
and it gives rise to the emergence of new peaks surrounded by those already present in DFT & TDDFT.
The combined effects of DFT+U and TDDFT+U proves to be almost a simple combination of their respective individual effects, as seen in EES with DFT+U & TDDFT+U, where excitation energies are globally shifted and some additional peaks emerge.
In Fig. 11 (also represented in Table 1), regardless of its flavour, TDDFT fails to capture the optically bright excitation at 2.85 eV observed in experiment, and this is consistent
with previous TDDFT studies using the LDA and PBE functionals.
Hybrid TDDFT using the B3LYP functional performs better than LDA or PBE
in this regard, surely due to its better (more spatially long-ranged)
description of exciton binding via its partial inclusion of the exact exchange interaction.
In Fig. 11, we see that DFT+U carries optically bright features to higher energies and dramatically changes the overall appearance of the spectrum.
In fact, DFT+U clearly worsens the agreement with
experimental excitation energies,
by pushing excitations within DFT & TDDFT to
higher energies such that the lowest optically bright excitation is carried to a position ∼1.8 eV higher energy compared to
that of DFT & TDDFT.
We find that TDDFT+U has a relatively minor effect on the optically bright excitations
when applied upon DFT (PBE), and no discernible effect when applied
upon DFT+U.
Thus, TDDFT+U does not mitigate the harmful effects of DFT+U on optically
bright excitations in this system.
TDA and RPA predict spectra in close mutual agreement, with slightly higher energies emerging within TDA for both spectra.
IV.2 Excitation energies and spectra of Ni(CO)4
The EES and ABS of Ni(CO)4 are presented in Fig. 12 and Fig. 13, respectively, for the first-principles Ueff=9.849 eV. Shown alongside, for comparison, are the corresponding spectra generated
using the experimental excitation energies and oscillator strengths extracted from Ref. 93.
In this molecule, due to its less-than-full 3d manifold
and hence increased 3d character of the valence-conduction
transition space,
we will see that TDDFT+U is rather more effective than it is in the case of
Ni(CN)42−.
However, it is still not enough to compensate for the inaccuracy that the
contemporary DFT+U potential introduces and, intriguingly,
DFT & TDDFT+U performs by far the best among the combinations
tested.
In Fig. 13
(also in Table 2, we observe that DFT & TDDFT overestimates the lowest optically bright excitation by ∼1.1 eV compared to in-vacuo
INDO/S
(the intermediate neglect of differential overlap model adapted
for spectroscopy) quantum-chemical calculations.
DFT+U worsens this over-estimation to
∼1.4 eV, while arguably also worsening the line-shape agreement.
TDDFT+U applied upon this (DFT+U & TDDFT+U)
makes relatively little difference,
and the effect of invoking the TDA is approximately that of a small,
rigid blue-shift.
It is difficult to make
a clear comparison against the large spread of experimental values, meanwhile.
The agreement between the peak positions and line-shapes
(we do not attempt to compare physical magnitudes here)
given by DFT & TDDFT+U and INDO/S, both for EES and ABS, is remarkable,
however,
with the first bright energy agreeing to ∼0.04 eV
(albeit with a splitting in INDO/S that is absent in TDDFT+U).
The ABS peak positions are also in reasonable agreement with some
of the experimental values given in Table 2, though again interpretation is challenging
here due to the spread of values.
We now digress to consider these results.
IV.3 The use of a single Hubbard U parameter
in DFT+U and TDDFT+U
The improvement of DFT & TDDFT by a first-principles Hubbard U
correction to the kernel but not to the potential,
if INDO/S can be taken
as a benchmark, may be understood as a possible consequence of the
following.
The Hubbard U parameter is a measure of spurious interaction,
one that is calculated as the derivative of an averaged potential which, in turn,
is a measure of the derivative of an energy.
On one hand, therefore, U is well
suited to measure the magnitude required for correction of
the interaction kernel.
On the other hand, it is not necessarily a good measure of the magnitude
required for correction of the Kohn-Sham potential.
More specifically, it has recently been shown by one of the present authors
that very different parameters U1 and U2 may be needed for the constant
and linear terms in the density, respectively, of the DFT+U corrective potential Moynihan et al. (2016).
Put another way, the linear and quadratic terms in Eq. 9
may benefit from different Ueff pre-factors.
Dubbed DFT+U1+U2, this generalization of DFT+U allows for
the approximate enforcement of Koopmans’ condition on the DFT+U
subspace, which is a condition that is
implied by the assumptions
under-pinning the calculation of U.
In other words, while the Hubbard U may successfully measure the
self-interaction strength, and possibly open the correct fundamental gap via the quadratic energy term,
a single parameter does not
carry enough information to correctly position the targeted subspace energetically with respect to the background (also known as bystander) states, a task for which the linear
term is better equipped.
Put yet another way, the double-counting
correction used in the derivation
of the contemporary DFT+U functional is arguably too simple,
for certain system types, and could gainfully
by given its own separate pre-multiplicative parameter.
The TDDFT+U kernel does not suffer from this complication,
however, since
only the usual parameter associated
with the quadratic energy term survives in the kernel.
In this sense, contemporary methods for
calculating a single U
parameter may actually be better suited to
TDDFT+U than to DFT+U.
This is reflected by the
apparently, paradoxically superior performance
of DFT & TDDFT+U over DFT+U & TDDFT+U
in the aforementioned system Ni(CO)4, albeit that this is
a rather extreme test of DFT+U insofar as that
the uncorrected PBE functional already performs well,
and that the relevant subspace is very far from half-filling.
Indeed, any ill-effects of conventional DFT+U on
the potential are expected to be most
strongly felt when applying DFT+U to spin-unpolarized 3d
spaces that are almost full (or empty) such as in
Ni(CO)4, since then the conduction (or valence)
band edge is of predominantly background-orbital
character.
The Kohn-Sham gap is neither of 3d→3d character
nor reliably determined by the familiar U in such cases.
A work-around alternative (albeit not equivalent) to DFT+U1+U2
may be the application of DFT+U to other orbital types, e.g.
O 2p, C 2p, and possibly Ni 4s, but this has not been explored in the
present work.
A complete counter-example to this, where DFT+U is very effective, is next provided by an
open-shell complex, where the Kohn-Sham gap is
strongly affected by a
varying Hubbard U parameter.
V First-principles spectra of a high-spin Cobalt-centred complex
CoL2Cl2 (L=2-aminopyrimidine: C4H5N3) is a Co-centred,
distorted pseudo-tetrahedral complex with two types of ligands, as illustrated in Fig. 14.
The central Co atom has a nominal charge of 2+, with a 3d sub-shell
containing 7 electrons.
Cl*-* is a π-donor weak-field ligand, which leads to a splitting of the 3d sub-shell of the Co atom into a high-spin configuration in a pseudo-tetrahedral symmetry Griffith (1964); Sundararajan et al. (2009).
In its high-spin configuration, the 3d orbitals at higher energies
contain unpaired electrons, resulting in a total
spin of 3 μB.
The fully and partially filled molecular orbitals at higher energies are predominantly hybrids comprised of Co 3d and Cl 3p orbitals.
Moreover, further splitting in the energy levels by 3d−3p hybridisation occurs by means of the distortion due to the tilted L-ligands.
The low-lying excitations are expected to have strong 3d→3d character in this molecule.
Experimental values for the low-lying, spin-allowed optically bright excitations of CoL2Cl2 are presented in Table 3.
Also provided are prior predictions from high-level quantum-chemistry methods, i.e., complete active space self-consistent field (CASSCF)
and CASSCF improved further by second-order N-electron valence perturbation theory (NEVPT2) Atanasov et al. (2011); Angeli et al. (2004, 2001, 2002), which were extracted from Ref. 107.
Our own TDDFT calculations invoke the TDA for this spin-polarized system,
due to technical limitations of the implementation.
Two different first-principles effective parameters
were tested in our DFT+U and TDDFT+U calculations, and
these were generated following the procedures
described in detail in Ref. 43.
Briefly, the like-spin Ueff=U−J
results from a treatment of the spin channels as forming an
effective 2-site model in the ‘scaled 2×2’ method,
and this is expected to yield results
(in this case 5.724 eV) comparable to those from
any correct method that separately calculates the Hubbard U
and Hund’s J.
The less canonical ‘averaged 1×1’ method
calculates the like-spin Ueff
as the average of the U parameters calculated individually for the two spin channels when decoupled (each forming part of the bath for the other),
and it may be a more reasonable
assumption when an explicit J correction term
is not used (as in the present work, where Ueff=3.798 eV).
In Fig. 15a, we see that DFT+U & TDDFT pushes
excitation features at lower energies higher, compared to DFT & TDDFT,
by ∼1.6−2.0 eV (∼1.0 eV) in the 2×2
(1×1) case.
In both cases an aggregate of
excitations forms at ∼2.8 eV, and in neither
case does DFT+U & TDDFT provide a promising agreement with
prior experiment or CASSCF-based results.
Meanwhile, the alternate combination, DFT & TDDFT+U,
which performed rather well in the case of Ni(CO)4,
was found to be
not at all viable here, for either Ueff value, as it gives rise
to unphysical, negative-valued excitation energies
(a single instability).
The interaction of DFT+U & TDDFT+U in this system is non-trivial, and
the net result cannot
be well described as a linear combination
(a cancellation) of the two method’s effects,
in general.
The linear combination picture holds to a greater degree for the
higher-valued, more canonical (2×2) prescription for Ueff, counter-intuitively.
With this, we find that uncorrected DFT & TDDFT does a better
job of reproducing the experimental absorption curve in
Fig. 15b, and that the absent low-lying, tightly-bound exciton
features predicted by CASSCF are no better recovered.
Here, referring to Fig. 15b, we emphasise that all curves
are independently normalised so that their maximum peak reaches a
value of unity, and that it is not necessarily the case that
DFT & TDDFT recovers the experimental maximum absorption cross-section by any means.
Conversely, with the lower-valued,
(1×1) prescription for Ueff, we find that the linear
combination picture breaks down completely.
With this Ueff, it appears that the effect of
DFT+U is insufficient to eradicate the strong
3d→3d character of the low-lying excitations.
Then, when TDDFT+U is applied on top of this, a very strong exciton
re-binding effect (of ∼2.0 eV) occurs, yielding a net
exciton binding effect of ∼1.0 eV with respect to DFT & TDDFT.
Ultimately, DFT+U & TDDFT+U within the 1×1 prescription for
Ueff does yield a group of tightly-bound ligand-field
excitations that can be said to be in qualitative agreement with the
CASSCF predictions of Ref. 107.
The accuracy improvements for lower-energy excitations offered by DFT+U & TDDFT+U are seen in Table 3.
Specifically, both DFT & TDDFT and DFT+U & TDDFT fail to capture the lowest three-fold degenerate excitation (highlighted with light pink) between ∼0.50−0.75 eV predicted at the level of CASSCF+NEWPT2).
Moreover, DFT+U & TDDFT also overestimates the second group of three-fold degenerate excitations (highlighted with light blue) at around ∼0.90−1.40 eV, either when compared against the experimental value of 1.12 eV or the CASSCF+NEWPT2 prediction of ∼0.95−1.13 eV.
DFT+U & TDDFT+U determines the lowest optically bright excitation energy with a relatively high accuracy at 0.56 eV, comparing to both CASSCF and CASSCF+NEWPT2.
Furthermore, it performs well by locating the second group of three-fold degenerate excitations (highlighted with light blue) at 0.90 eV and 1.02 eV.
However, DFT+U & TDDFT performs better, without a doubt, for the third
group of three-fold degenerate excitations (highlighted with light purple) at
2.45 eV, when comparing to the experimental value.
Overall, we can conclude that first-principles (1×1 prescription)
DFT+U & TDDFT+U performs better for low-lying excitations than DFT & TDDFT,
but this comes at the expense of completely removing the
prominent absorption peak at ∼2.0 eV where experiment and
DFT & TDDFT agree.
None of the available methods (including CASSCF),
therefore, offer reliable correction of the spectra
for both 3d→3d and
higher-energy excitations, and this
is as expected given the spatially localized nature of Hubbard U
corrections when applied to metal 3d orbitals only.
VI Conclusion
In this work, we carried out a systematic investigation of the extension of
Hubbard U corrected approximate Kohn-Sham DFT to the excited-state
regime, namely TDDFT+U.
For this, a linear-scaling, linear-response implementation of TDDFT+U
was developed within the ONETEP code,
by combining existing linear-scaling DFT+U O’Regan et al. (2012); Han et al. (2006),
conduction-band optimization Ratcliff et al. (2011), and beyond Tamm-Dancoff
TDDFT Zuehlsdorff et al. (2013, 2015, 2016) methods.
Our implementation has allowed us to decouple and
analyse the separate and combined effects of Hubbard U
correction at the DFT (potential) and TDDFT (kernel) levels,
offering insights into the performance and potential range
of useful applicability of TDDFT+U.
A four-level toy model has proved invaluable
to our interpretation of TDDFT+U and
the numerical results that support this picture,
particularly in two representative
low-spin (spin-unpolarised but non-isoelectronic)
Ni-centred complexes.
In these systems, we first treated the
Hubbard U as a free parameter in order
to understand in detail the exciton binding effect of TDDFT+U, as well
as the tendency for the effects of DFT+U
and TDDFT+U to approximately cancel.
We also analysed in detail the differing effects of Hubbard U
on TDDFT depending on whether the Tamm-Dancoff approximation
is invoked.
Including also a challenging Co-centred open-shell, high spin coordination complex,
we calculated first-principles Hubbard U and Hund’s J
parameters for all three systems, following the spin-polarised,
minimum-tracking Moynihan et al. (2017) linear-response approach introduced
in Ref. 43.
This has enabled us to generate fully first-principles
excitation and absorption spectra for each of these
elusive systems and to compare with prior experimental and
quantum chemical findings.
Physically, our analysis shows that TDDFT+U can be thought of as a
self-interaction correction for excitons, acting to enhance the exciton
binding.
Indeed, quite apart from TDDFT+U being mandated in principle when TDDFT is
applied upon a DFT+U Kohn-Sham eigensystem, we find that TDDFT+U can be very
effective in re-binding well-defined strongly-localized, optically dark ligand-field
excitations.
The Hubbard U dependence of this re-binding is illustrated nicely, we
think, in Fig. 5c.
Our study has identified examples of such ligand-field excitations that are
predicted at low energies by quantum-chemistry methods but
pushed to unrealistically high energies by first-principles DFT+U.
TDDFT+U can address this effectively, to some extent, but
only if the localized character
of those excitations has not already been eradicated by DFT+U, however,
as illustrated in Fig. 15a.
In general, while DFT+U shifts excitation energies of transitions into, out of,
and within the targeted localised subspace by modifying
the underlying Kohn-Sham energy levels in
proportion to the effective Hubbard U parameter,
approximately speaking,
TDDFT+U only directly affects transitions within that subspace.
This gives rise to an incomplete cancellation of the effects of
DFT+U and TDDFT+U and
as a result, we conclude that while the combination of DFT+U and TDDFT+U may
often give rise to something of a linear combination of the two method’s
effect, the interaction between them may also be
non-trivial, with multiple U-dependence regimes potentially emerging.
Existing approaches for the calculating the adiabatic limit of
the Hubbard U and Hund’s J within DFT (or more precisely
generalised Kohn-Sham DFT, in practice), such as linear-response
method, already calculate the necessary
parameters for TDDFT+U by construction.
Indeed, our results suggest that these parameters may be more suited
to TDDFT+U than to DFT+U, in the sense that U
(and J) exist at the same energy-derivative order as the kernel correction
fUσσ′, whereas the DFT+U correction to the potential
retains a somewhat arbitrary constant (in the sense that a choice
of double-counting correction must be made).
Furthermore, our results add to the growing body of literature that suggests that
DFT+U should be used with caution on closed-shell, or more generally
low-spin systems, as discussed in Ref. 43
and references therein.
Our findings on the closed-shell complex Ni(CO)4, for example,
where DFT & TDDFT+U performs rather well when judged against
the INDO/S quantum chemistry method
(see third panel of Fig. 13), suggest a basic failure of the
DFT+U corrective potential in combination with the
first-principles Ueff=U−J.
An interesting avenue for future investigation in
problematic systems such as those studies is the use of a
second Hubbard U parameter to enforce Koopmans’
condition to the targeted subspace Moynihan et al. (2016),
as discussed in Section IV.3.
This idea effectively fixes the arbitrary constant in DFT+U,
or locates the double-counting correction from first principles,
but its effect in non-trivial systems is yet to be investigated.
Overall, notwithstanding, a picture emerges in the present work whereby
the application of Hubbard U correction to a single localized subspace
alone (with first-principles parameters Linscott et al. (2018))
may be advantageous
and expedient for the qualitative description of
optically dark 3d→3d excitations that are difficult to otherwise
recover.
This description can come, however, at the expense of considerably worsening
the description of less localized excitations that are well described by standard,
semi-local approximations to TDDFT.
Further research is warranted, therefore, on generalizations to the
contemporary DFT+U functional such as to incorporate
further chemical information.
More basically, perhaps,
but no less interestingly, more research is needed on the
effects of DFT+U,
DFT+U+J Himmetoglu et al. (2011),
DFT+U+V Jr and Cococcioni (2010) (and
their potential respective
TDDFT+U extensions) to more
delocalised subspaces centred on ligand atoms
(see for example the oxygen
2p treatment in Ref. 43)
or even bond-centred ones.
VII Acknowledgements
We gratefully acknowledge the support of
Trinity College Dublin’s Studentship Award
and School of Physics.
The authors also acknowledge the DJEI/DES/SFI/HEA Irish Centre for High-End Computing (ICHEC) for the provision of computational facilities and support.
We also acknowledge Trinity Centre for High Performance Computing
(Trinity Research IT)
and Science Foundation Ireland, for the maintenance and
funding, respectively, of the Lonsdale and Boyle clusters on which
further calculations were performed.
Appendix A Computational details
First-principles simulations were performed using
our implementation of the TDDFT+U
method in the ONETEP linear-scaling package Skylaris et al. (2005); Haynes et al. (2006); O’Regan et al. (2012).
All calculations used the Perdew-Burke-Ernzerhof (PBE) Perdew et al. (1996)
generalized gradient approximation as the underlying
exchange-correlation functional.
Norm-conserving scalar-relativistic PBE pseudo-potentials were generated
in-house for neutral Ni, Cl, O, C, N, H,
and Co2+ using the OPIUM code opi .
Ground-state simulations are referred to here
as single-point (SP), and the subsequent
procedure of variationally optimising the second set of NGWFs to represent the unoccupied manifold Ratcliff et al. (2011) is referred as conduction (COND).
Initial ionic geometries were adopted from a prior first-principles study Demuynck et al. (1971) in the case of Ni(CN)42−, and from experimental data Hedberg et al. (1979) in the case of Ni(CO)4.
These molecular geometries were optimized iteratively
until they fulfilled three convergence criteria: on the maximum atomic displacements (0.005 a0), total energy per atom
(10−6 Ha), and total atomic force (0.002 Ha/a0), by means of the Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm Hine et al. (2011); Ruiz-Serrano et al. (2012).
In the case of the CoL2Cl2, the molecular geometry was directly adopted from Ref. 115 for the sake of preserving with comparability of the spectra of Ref. 107, which use the same geometry.
The molecules were then positioned into smaller cuboidal simulation boxes centred on their respective metallic atoms, with the available minimum dimensions needed to satisfy the requirements of the Martyna-Tuckeman periodic boundary correction (PBC), which was applied with its dimensionless parameter set to 7 as recommended in Ref. 116.
A series of convergence tests were performed to
safeguard the quality excited-state simulations, while maintaining a reasonable computational cost at the SP, COND and TDDFT levels
(recalling that the effective U is treated as a parameter, which significantly
multiplies the total computational demand of the study).
The resulting common set of parameters used in this study is summarized in Table 4.
The effective plane-wave kinetic energy cut-off (Ecut) and the cut-off radius (RNGWFs) of the variationally-optimized
nonorthogonal generalized Wannier functions
NGWFs, a minimal basis generated by ONETEP,
were converged at values of 1200 eV and 12 a0, respectively,
yielding a energy error per atom within 1 meV in SP calculations.
The value of RNGWF was separately tested in COND calculations
and found to be adequate for describing the virtual orbital eigen-energies. A total of 9(18) spin-degenerate NGWFs
were used for Ni atoms in order to complete the period up to Kr, and a total of 4 NGWFs for each of C, O and N were used to complete the period up to Ar, were optimized at the SP (COND) level in our Ni-centered complexes, whereas
for the Co-centred complex 9 (18), 4 (13), 4 (8), and 1 (2) NGWFs were variationally optimized for Co, Cl, (C,N), and H atoms during SP (COND) simulations
As CoL2Cl2 is an open-shell system, spin-polarized calculations were performed with a fixed total spin of 3 μB,
and the initial configuration of Co for the pseudo-atomic solver
(which effects both the NGWF initial guess and the 3d pseudo-orbitals defining the DFT+U
subspace) was set to the theoretical high-spin
configuration of [Ar]4s03d7, with a 3 μB total spin.
The occupied-unoccupied Kohn-Sham eigenvector product spaces were constructed by using full valence manifolds, which are represented by 24 and 25 spin-degenerate NGWFs in Ni(CN)42− and Ni(CO)4, respectively, and 49 and 46 NGWFs for spin-up and spin-down, respectively, in CoL2Cl2.
For the conduction manifolds, 20 (10 per spin channel), 16 (8 per spin channel) and 11
(4 for up and 7 for down) KS conduction orbitals were optimized in Ni(CN)42-, Ni(CO)4, and CoL2Cl2, respectively.
These parameters were selected on the basis of KS eigenvalues, providing
sufficiently many bound states for the targeted spectral range in TDDFT calculations.
The first 50 singlet excitations for Ni-centered complexes and 20 singlet excitations for CoL2Cl2 were calculated by variational minimization,
within the larger valence-conduction product space spanned by the
optimized NGWF basis .
We do not place a strong emphasis on the higher-energy
excitations shown in our plots, being more interested and
confident in the lower-energy excitations affected
by the Hubbard U correction.
In particular, in many of our figures the EES and ABS appear gapped at
high energy, but this is nothing more than
an artefact of the limited number
of excitations calculated.
Appendix B First-principles calculation of Hubbard U and J
parameters using the minimum-tracking linear-response
method
The efficiency and robustness of the DFT+U(+J) method is essentially dependent on the determination of the Hubbard parameters.
A common approach is to use linear-response to determine them Pickett et al. (1998); Cococcioni and de Gironcoli (2005).
In this work, we employ the recently-introduced minimum-tracking variant of linear-response as implemented in the ONETEP code Moynihan et al. (2017), and in particular, its spin-polarized extension introduced in Ref. 43.
In this, the ‘scaled 2×2’ method can be used to
evaluate the Hubbard U, Hund’s J, and effective Hubbard
U parameter (Ueff=U−J) for all three systems using the formulae
[TABLE]
where
[TABLE]
The spin-dependent
interaction strengths fσσ′
are calculated by incrementally varying subspace-uniform
perturbatimg potentials δvextσ,
relaxing fully to the ground-state on each step, and then
measuring the resulting small changes
in the subspace occupancies
nσ and subspace-averaged Kohn-Sham
potentials vKSσ.
The projected interacting response matrices are given by
χσσ′=dnσ/dvextσ′.
When the interaction strengths fσσ′
are calculated using a 2×2 matrix equation
indexed by spin, we arrive at the ‘scaled 2×2’ model,
which reproduces conventional formulae for U and J.
Indeed, for spin-unpolarized systems such as the Ni-centered complexes
studied in this work, λU=1 and λJ=−1,
and as a result we have U=(fσσˉ+fσσ)/2,
J=(fσσˉ−fσσ)/2,
and, simply but reassuringly, Ueff=fσσ.
When spin-off-diagonal elements are neglected, instead,
we have the ‘averaged 1×1’ model, in which
Ueff=(U↑+U↓)/2,
where Uσ=d(vKSσ−vextσ)/dnσ.
This model effectively decouples the spin
populations into distinct sites, reflecting the
form of the canonical DFT+U functional.
Each spin channel, for a given localized subspace, then forms part of the screening bath for the other, and
the effects of Hund’s J are then already
incorporated into Ueff at an approximate level.
In practice, a discrete logarithmic grid of
perturbation strengths,
{−0.10,−0.01,0.00,0.01,0.10} eV,
was used in this work
to calculate the U and J parameters, resulting
in excellent linear fits.
For the spin-unpolarized Ni-centred complexes, it was
necessary only to perturb one spin channel, since half
of the spin-indexed matrix elements could be filled
using symmetry.
The resulting parameters are summarized in Table 5.
As CoL2Cl2 is a spin-polarized system, the responses of each spin channel were measured by perturbing the respective spin channels, separately, one at a time.
The resulting first-principles parameters for the Co 3d subspace are summarised in Table 6.

 Figure 1
Figure 1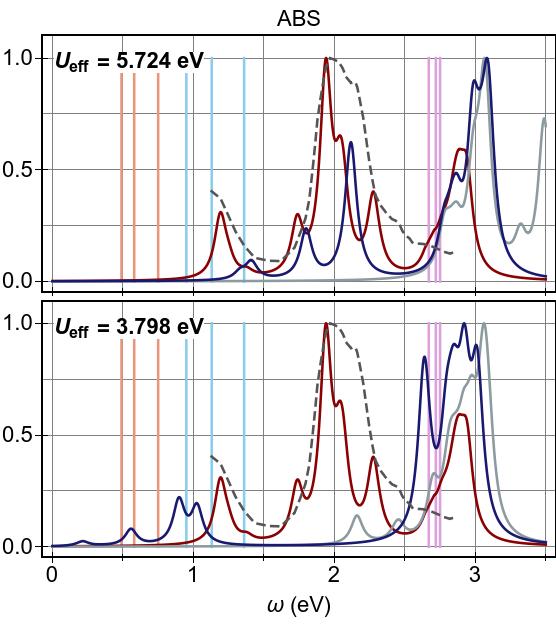 Figure 2
Figure 2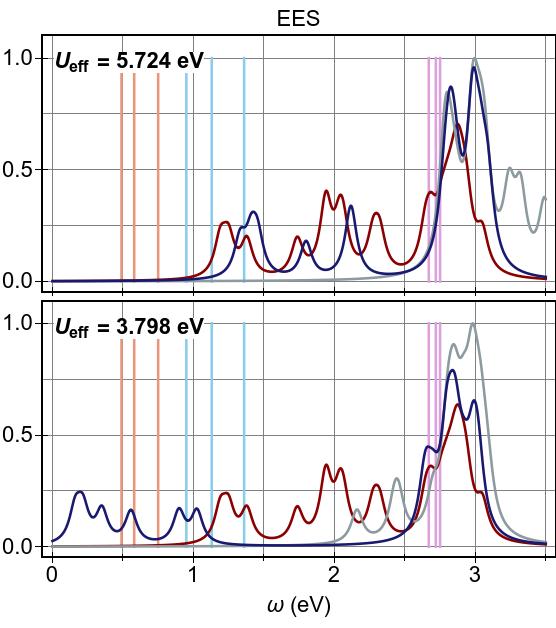 Figure 3
Figure 3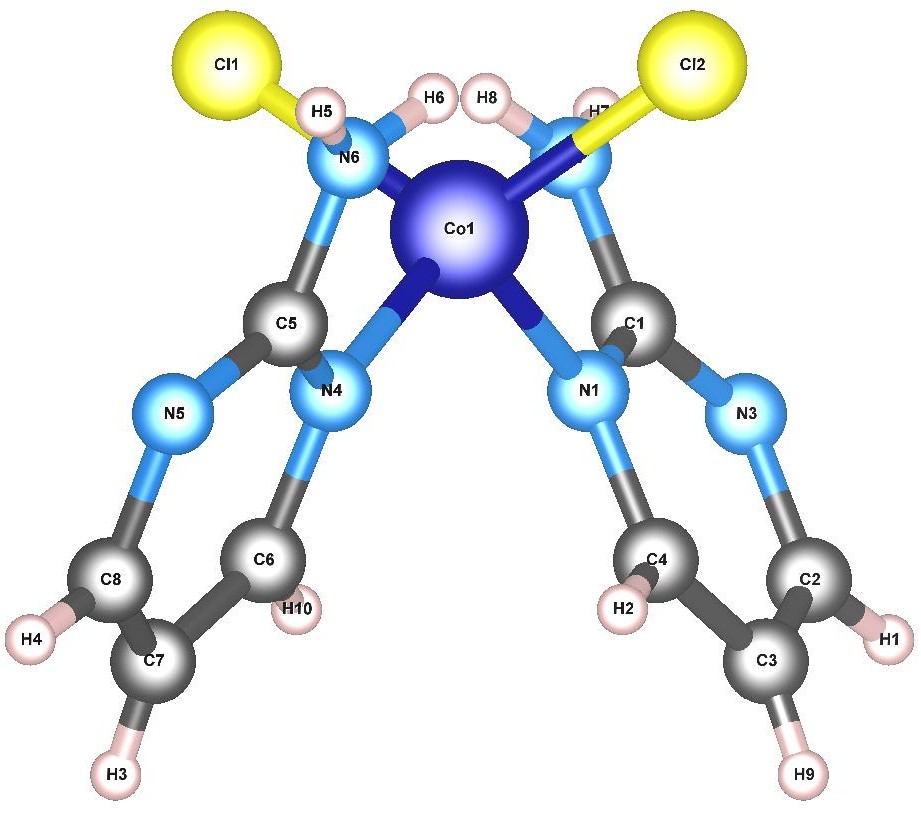 Figure 4
Figure 4 Figure 5
Figure 5 Figure 6
Figure 6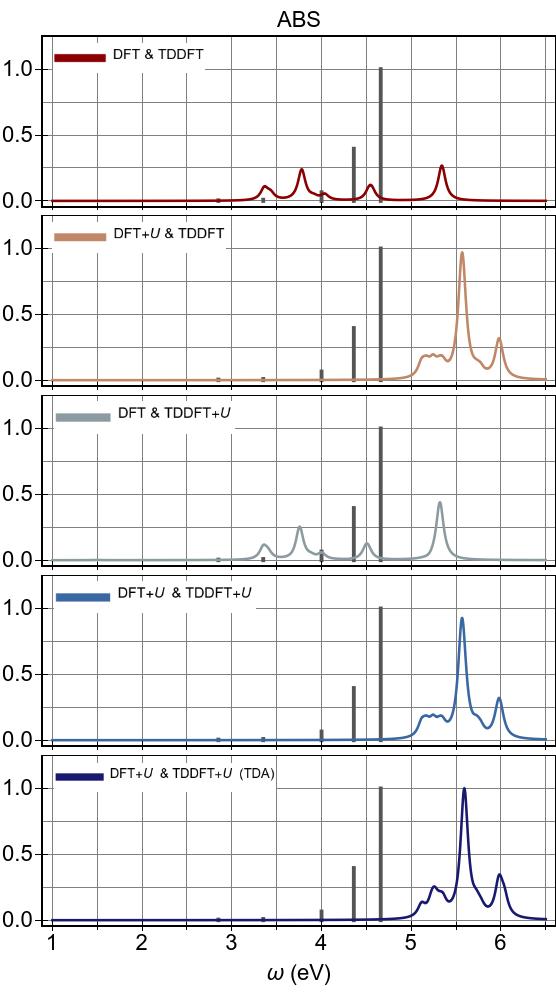 Figure 7
Figure 7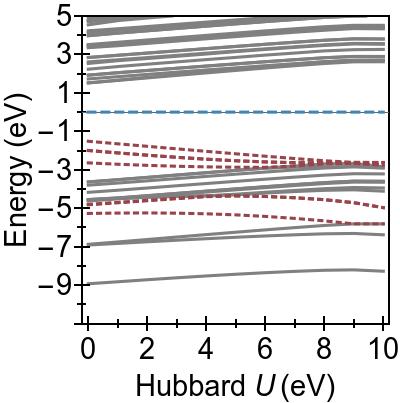 Figure 8
Figure 8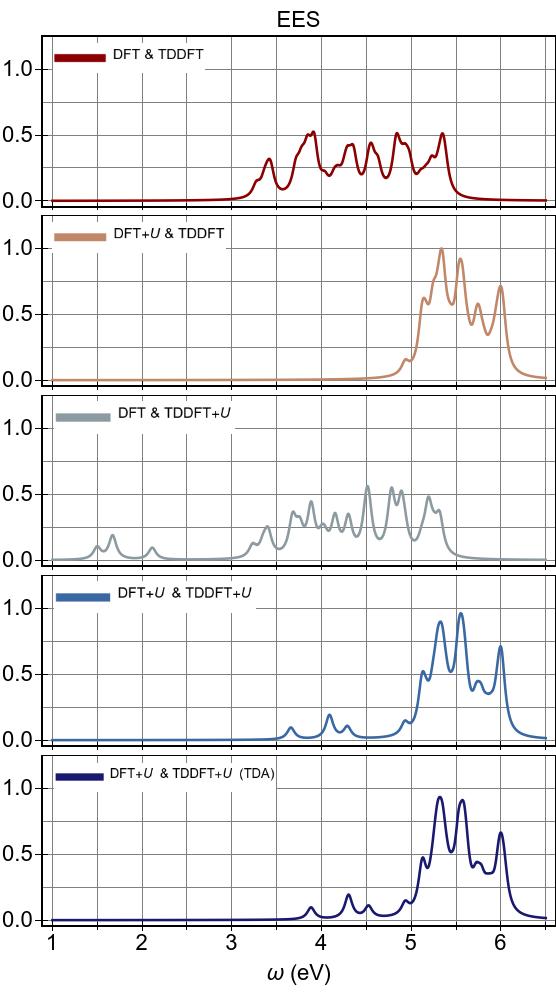 Figure 9
Figure 9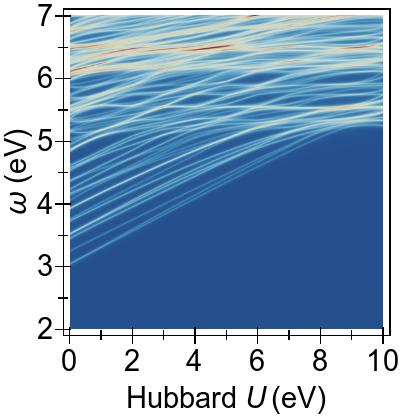 Figure 10
Figure 10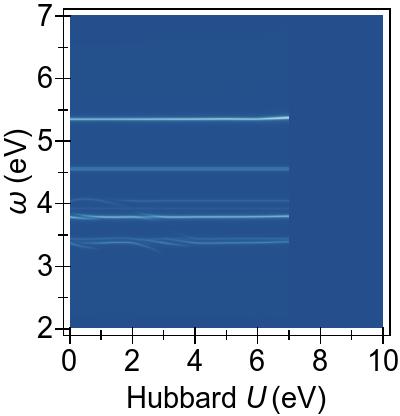 Figure 11
Figure 11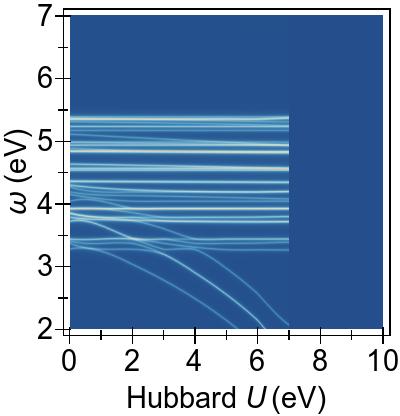 Figure 12
Figure 12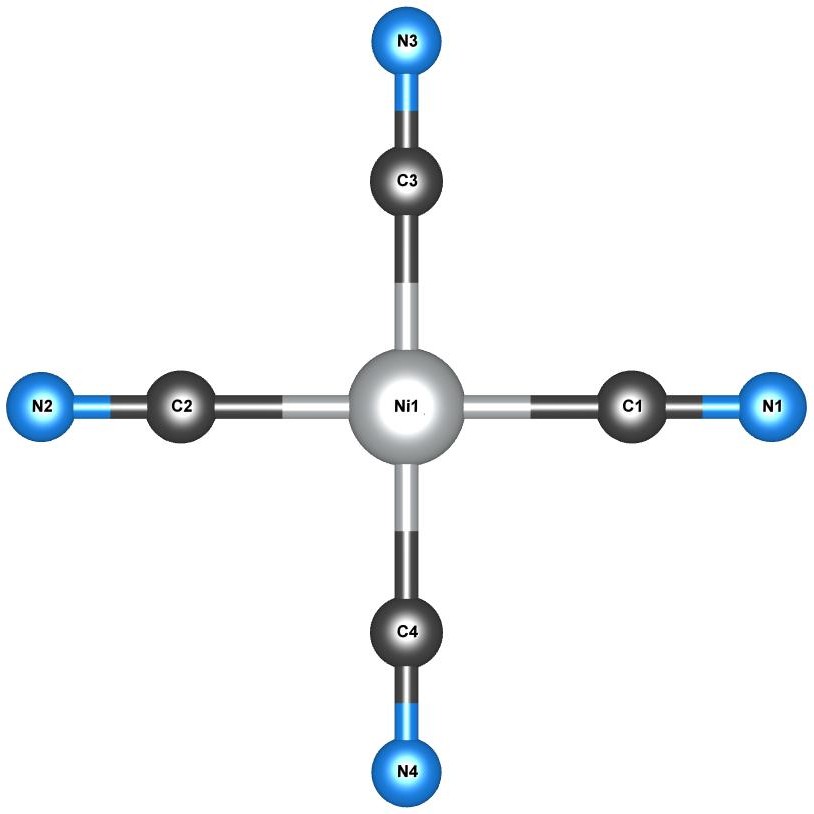 Figure 13
Figure 13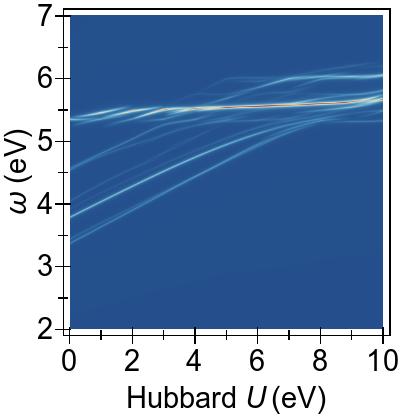 Figure 14
Figure 14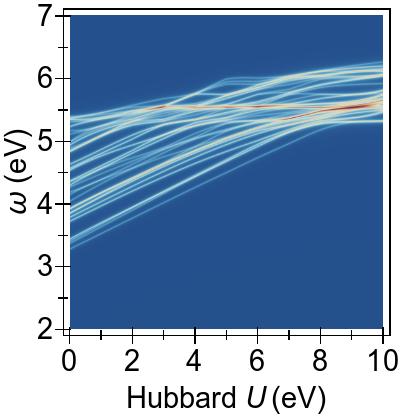 Figure 15
Figure 15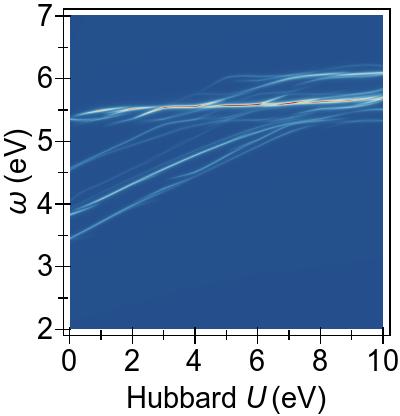 Figure 16
Figure 16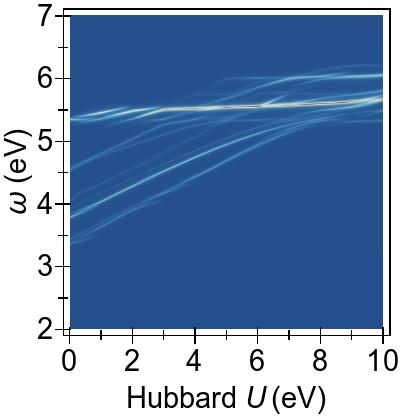 Figure 17
Figure 17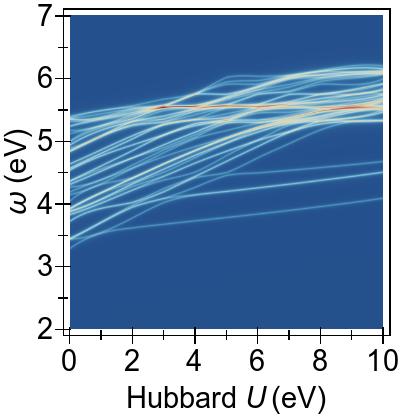 Figure 18
Figure 18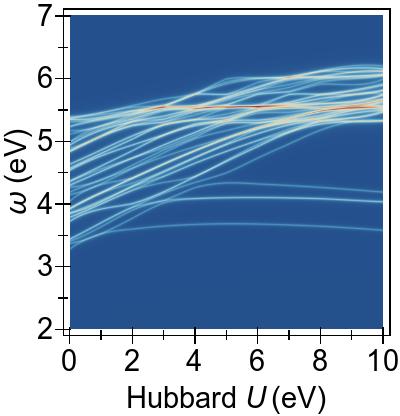 Figure 19
Figure 19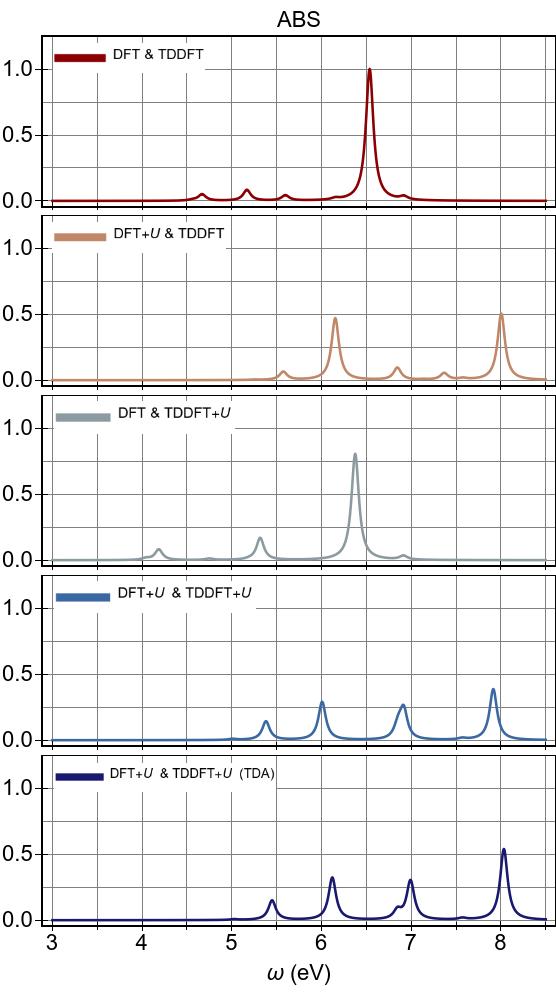 Figure 20
Figure 20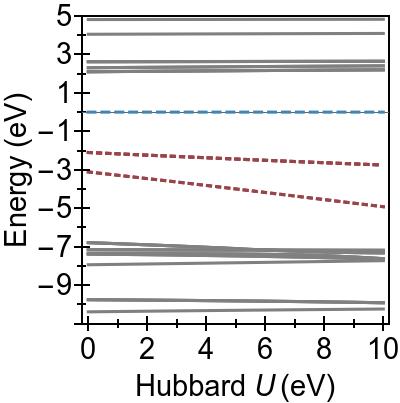 Figure 21
Figure 21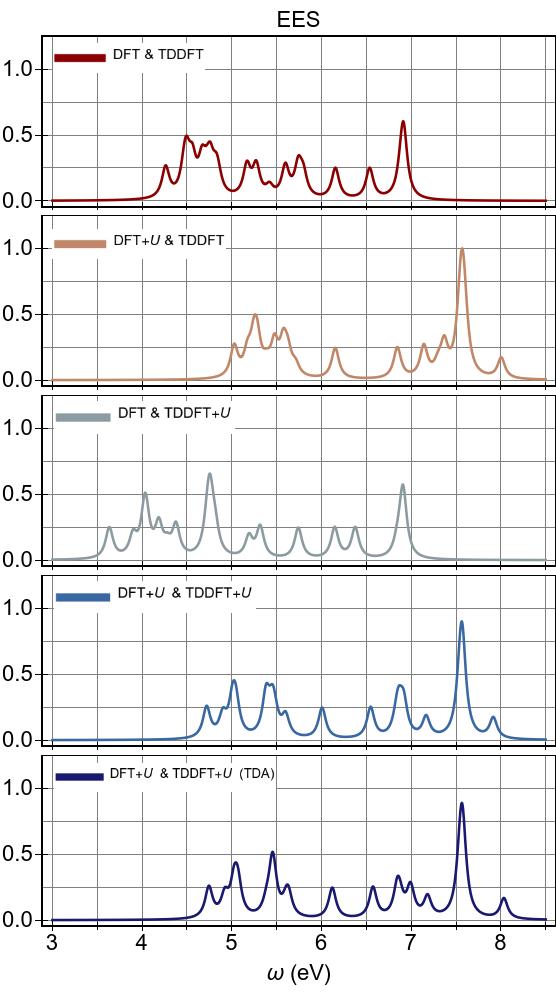 Figure 22
Figure 22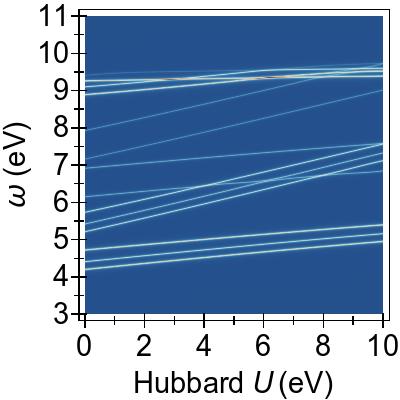 Figure 23
Figure 23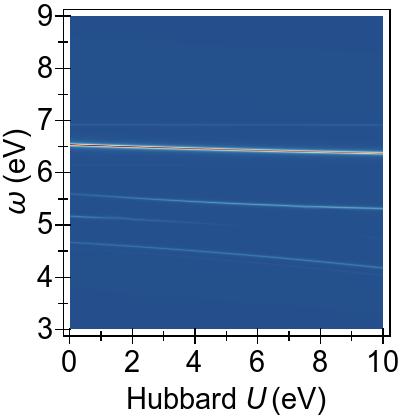 Figure 24
Figure 24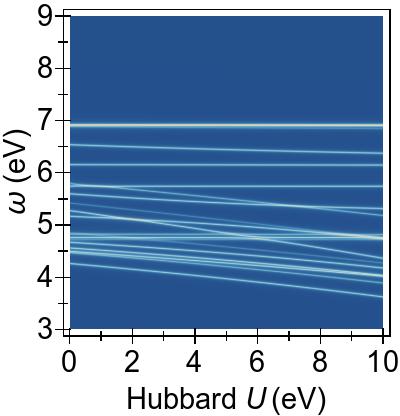 Figure 25
Figure 25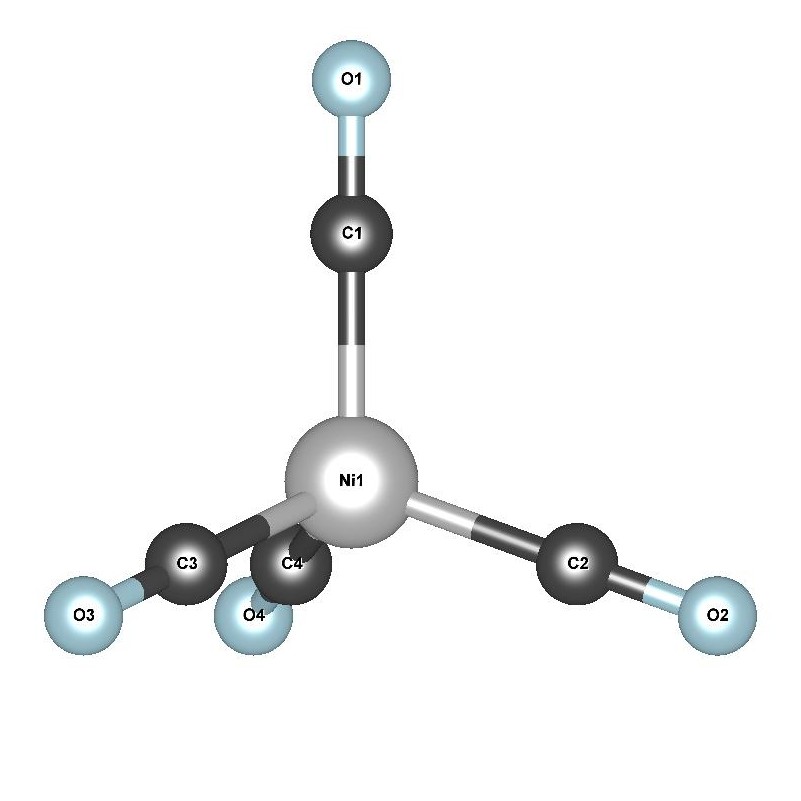 Figure 26
Figure 26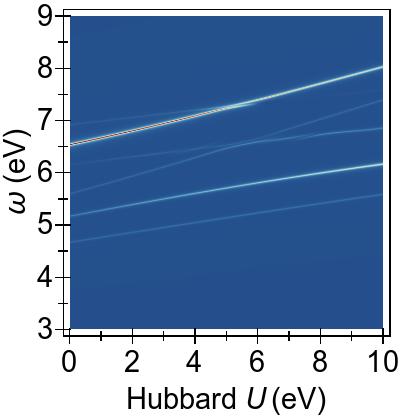 Figure 27
Figure 27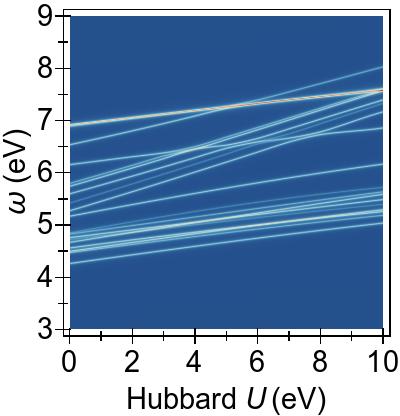 Figure 28
Figure 28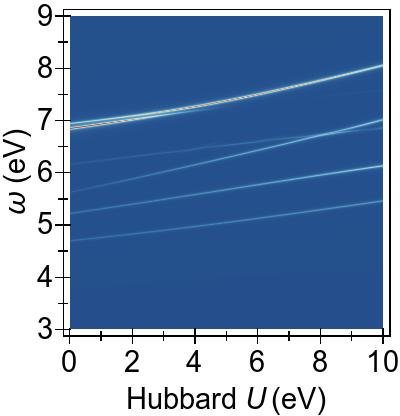 Figure 29
Figure 29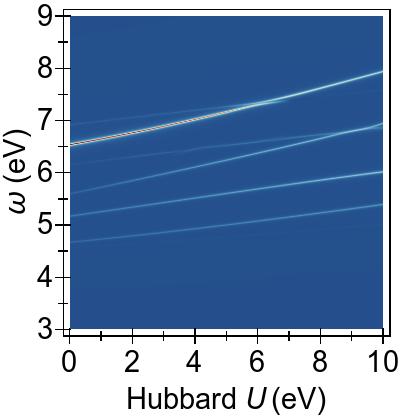 Figure 30
Figure 30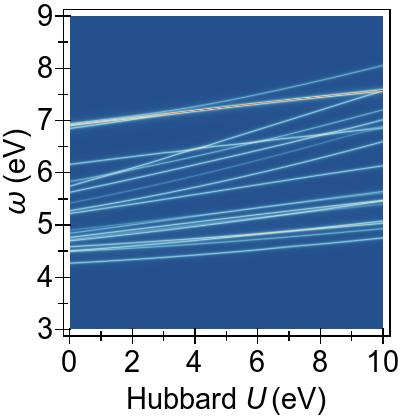 Figure 31
Figure 31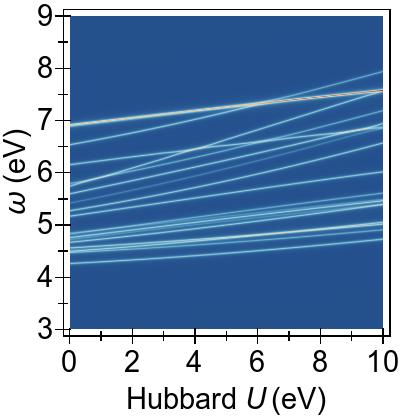 Figure 32
Figure 32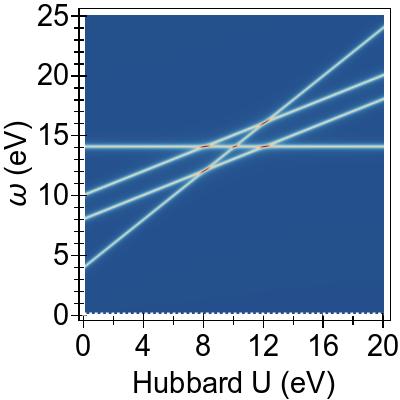 Figure 33
Figure 33 Figure 34
Figure 34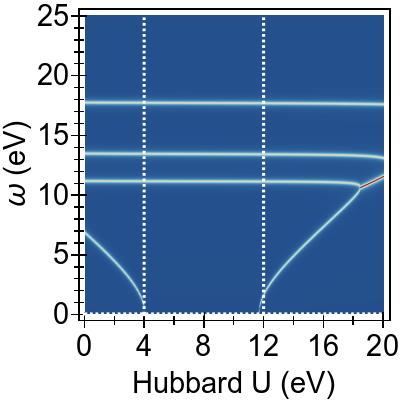 Figure 35
Figure 35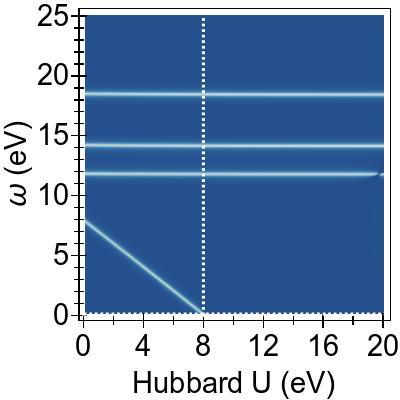 Figure 36
Figure 36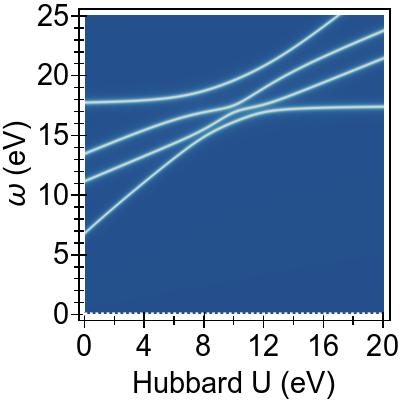 Figure 37
Figure 37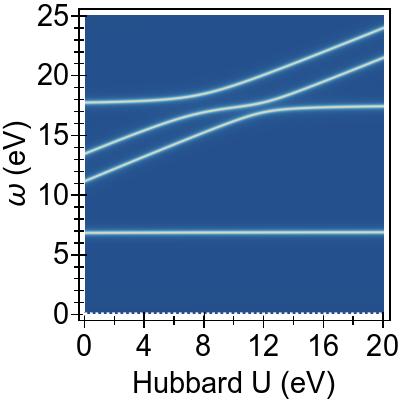 Figure 38
Figure 38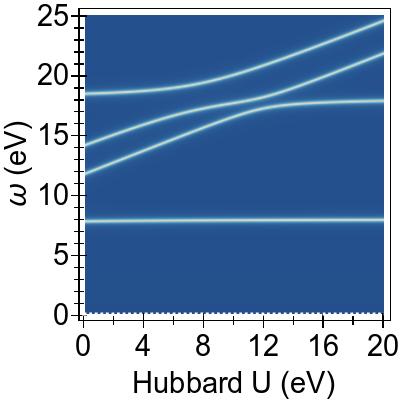 Figure 39
Figure 39