Effects of Hubbard term correction on the structural parameters and electronic properties of wurtzite Zn
E. S. Goh, J. W. Mah, T. L. Yoon

TL;DR
This study investigates how Hubbard term corrections affect the structural and electronic properties of wurtzite ZnO, highlighting the importance of parameter selection for accurate predictions.
Contribution
It demonstrates that combining Hubbard corrections for Zn 3d and O 2p states can accurately reproduce the experimental band gap of ZnO, but may not improve vibrational property predictions.
Findings
Hubbard corrections widen the band gap to match experiments.
Lattice constants are underestimated with Hubbard corrections.
Vibrational frequencies do not match experimental values despite electronic improvements.
Abstract
The effects of including the Hubbard on-site Coulombic correction to the structural parameters and valence energy states of wurtzite ZnO was explored. Due to the changes in the structural parameters caused by correction of hybridization between Zn d states and O p states, suitable parameters of Hubbard terms have to be determined for an accurate prediction of ZnO properties. Using the LDA+ method by applying Hubbard corrections to Zn 3d states and to O 2p states, the lattice constants were underestimated for all tested Hubbard parameters. The combination of both and correction terms managed to widen the band gap of wurtzite ZnO to the experimental value. Pairs of and parameters with the correct positioning of d-band and accurate bandwidths were selected, in addition to predicting an accurate band gap value. Inspection of vibrational properties,…
Click any figure to enlarge with its caption.
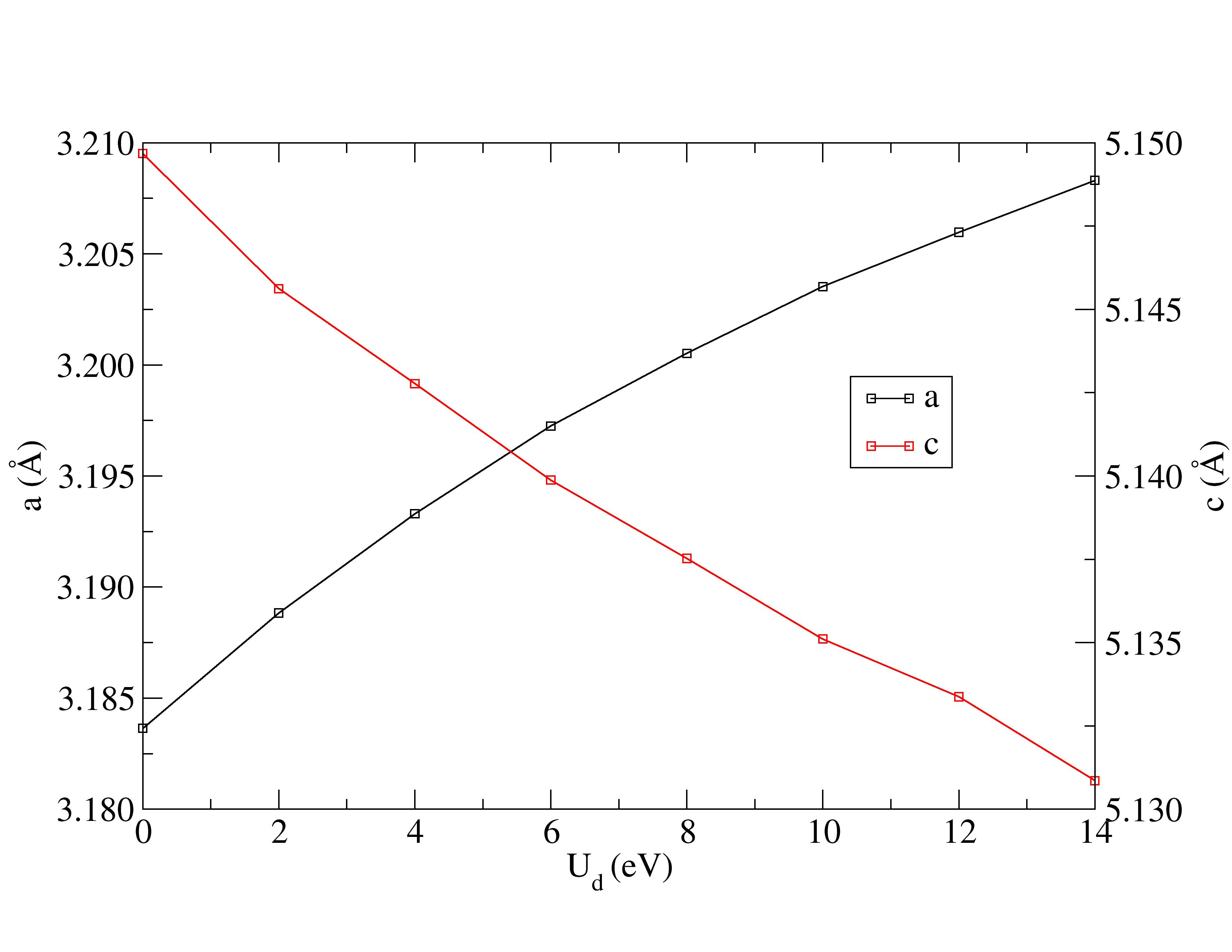 Figure 1
Figure 1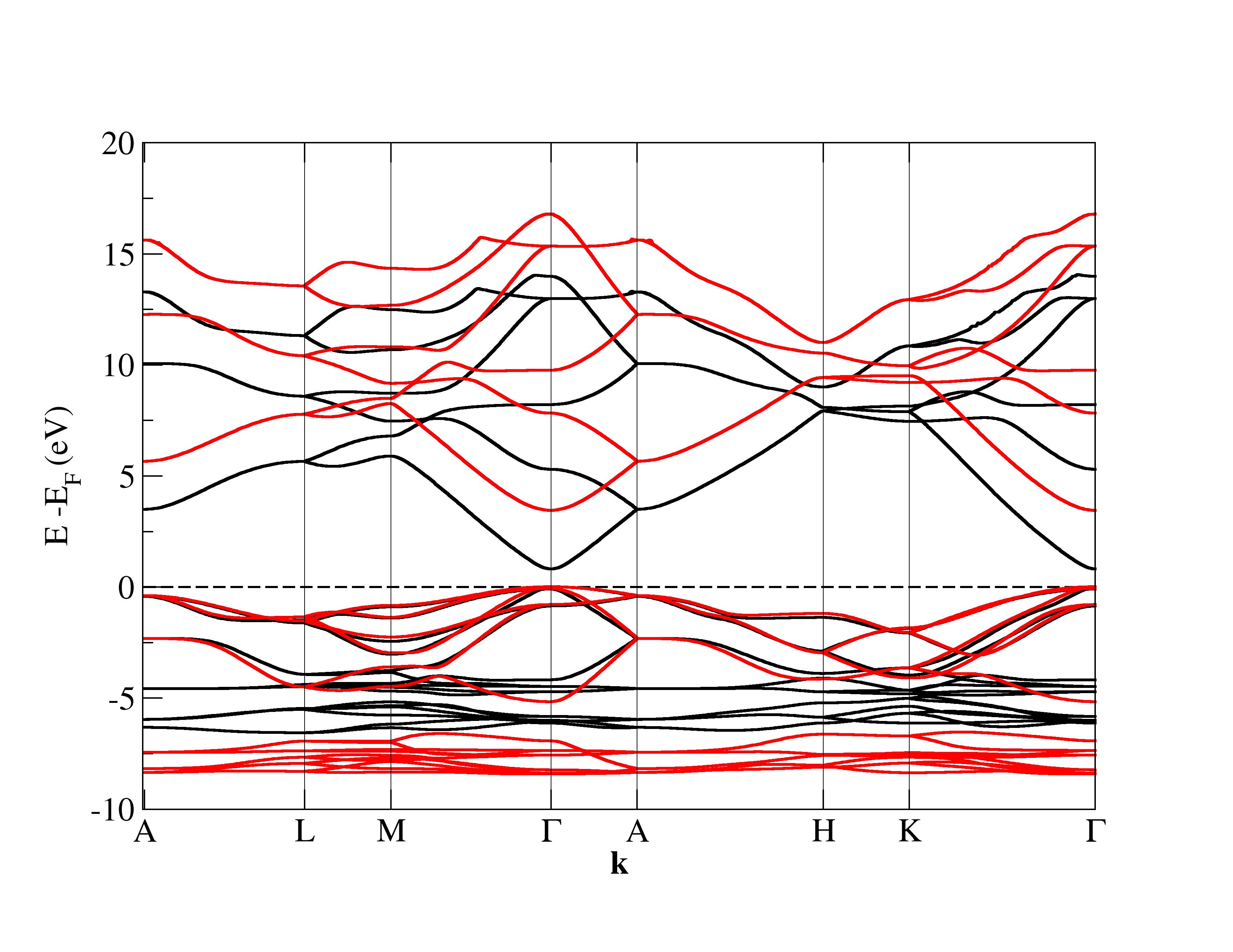 Figure 2
Figure 2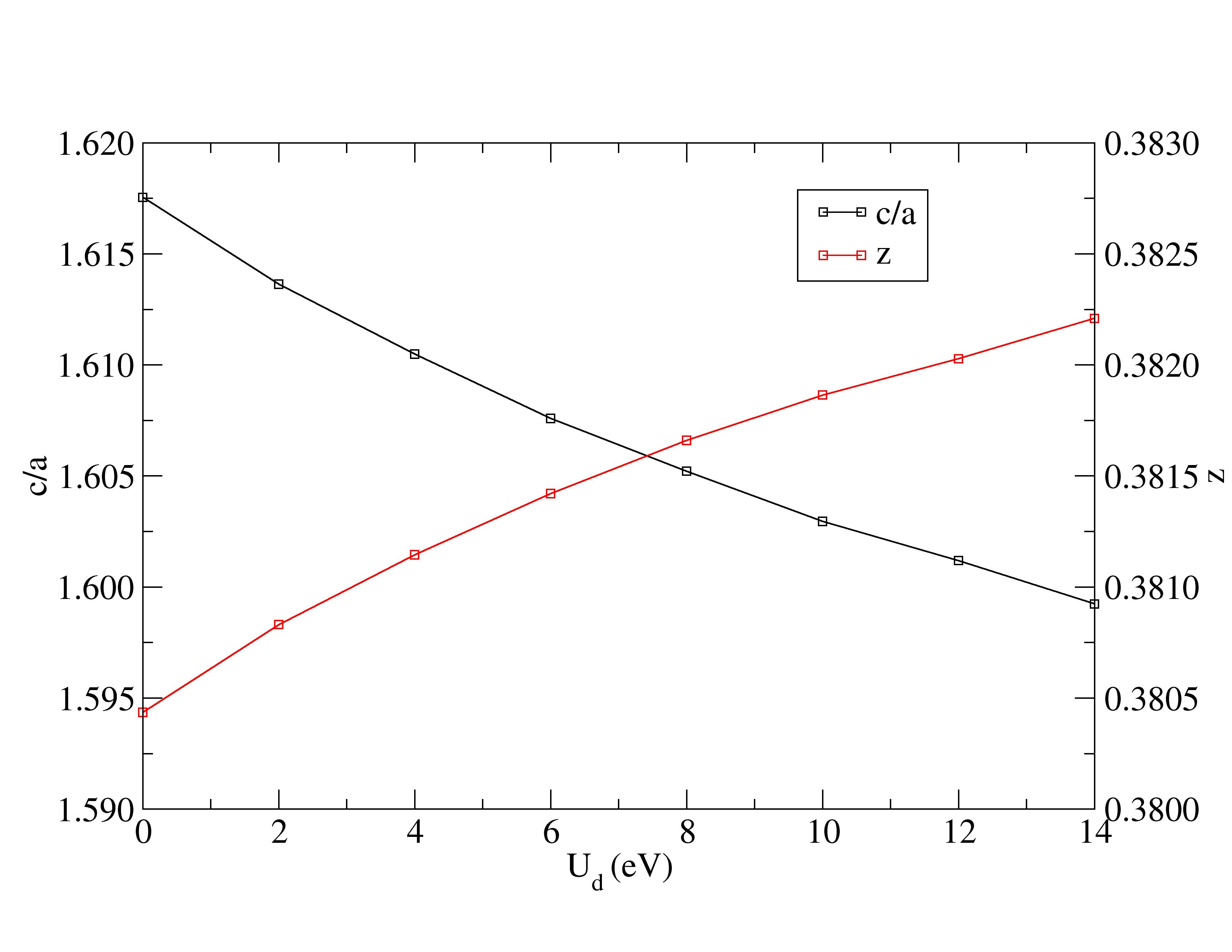 Figure 3
Figure 3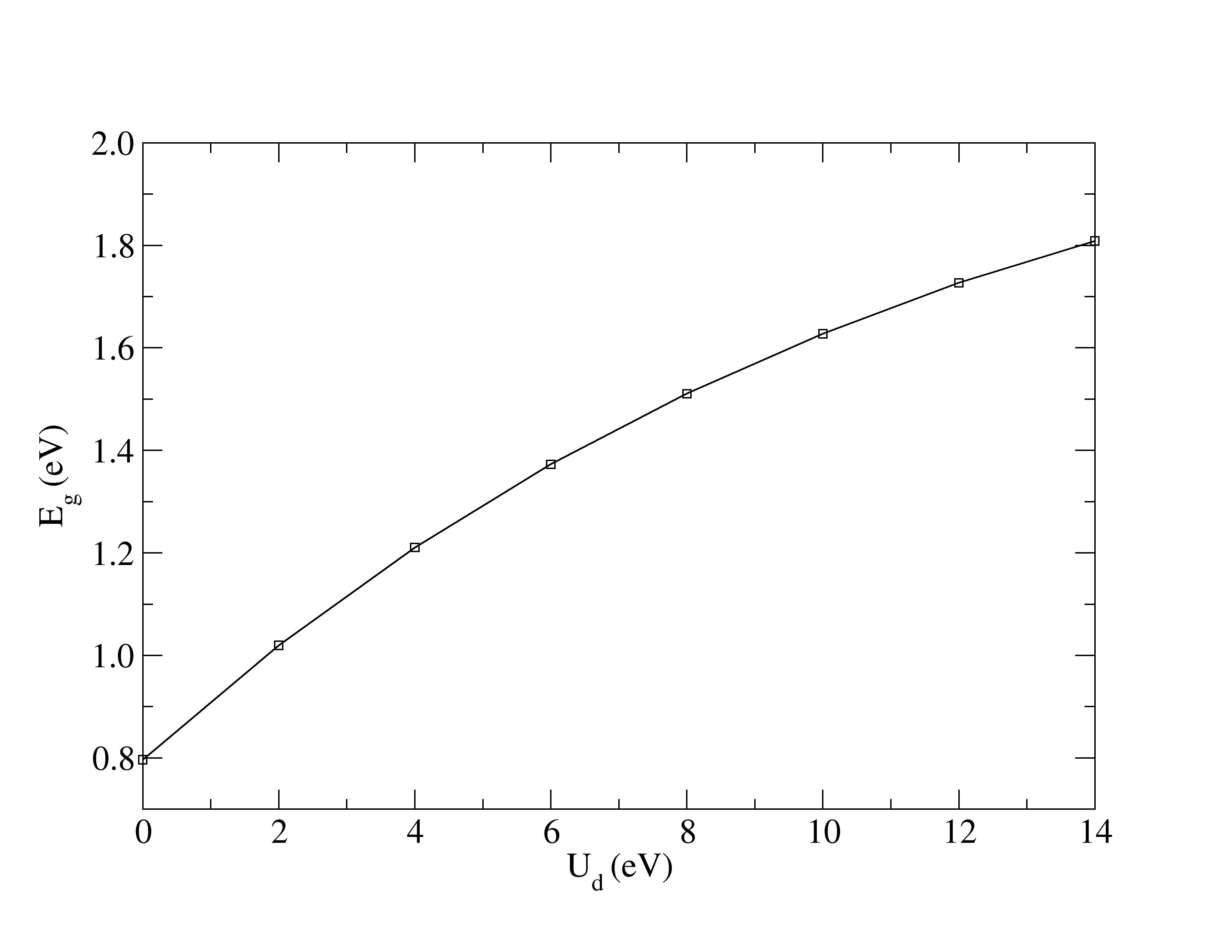 Figure 4
Figure 4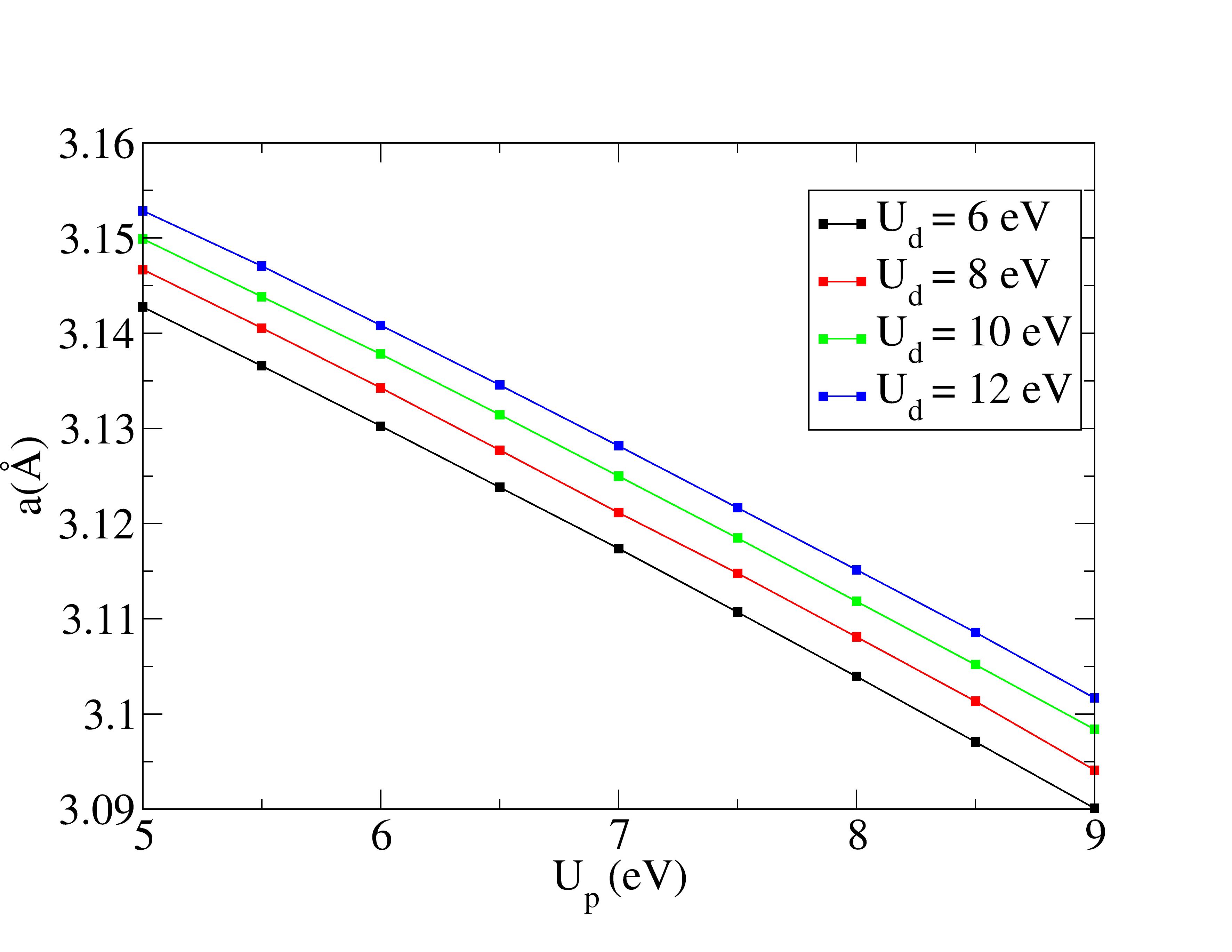 Figure 5
Figure 5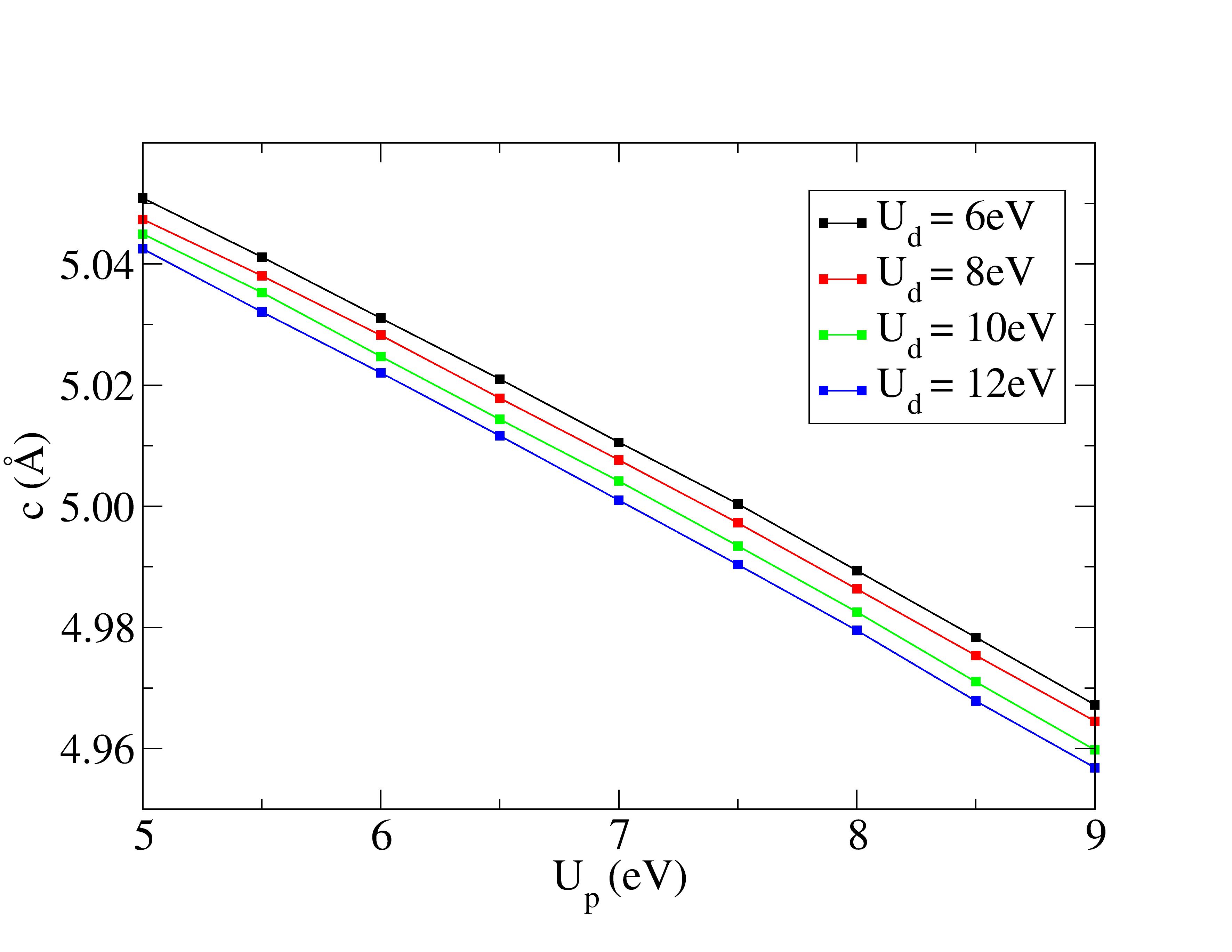 Figure 6
Figure 6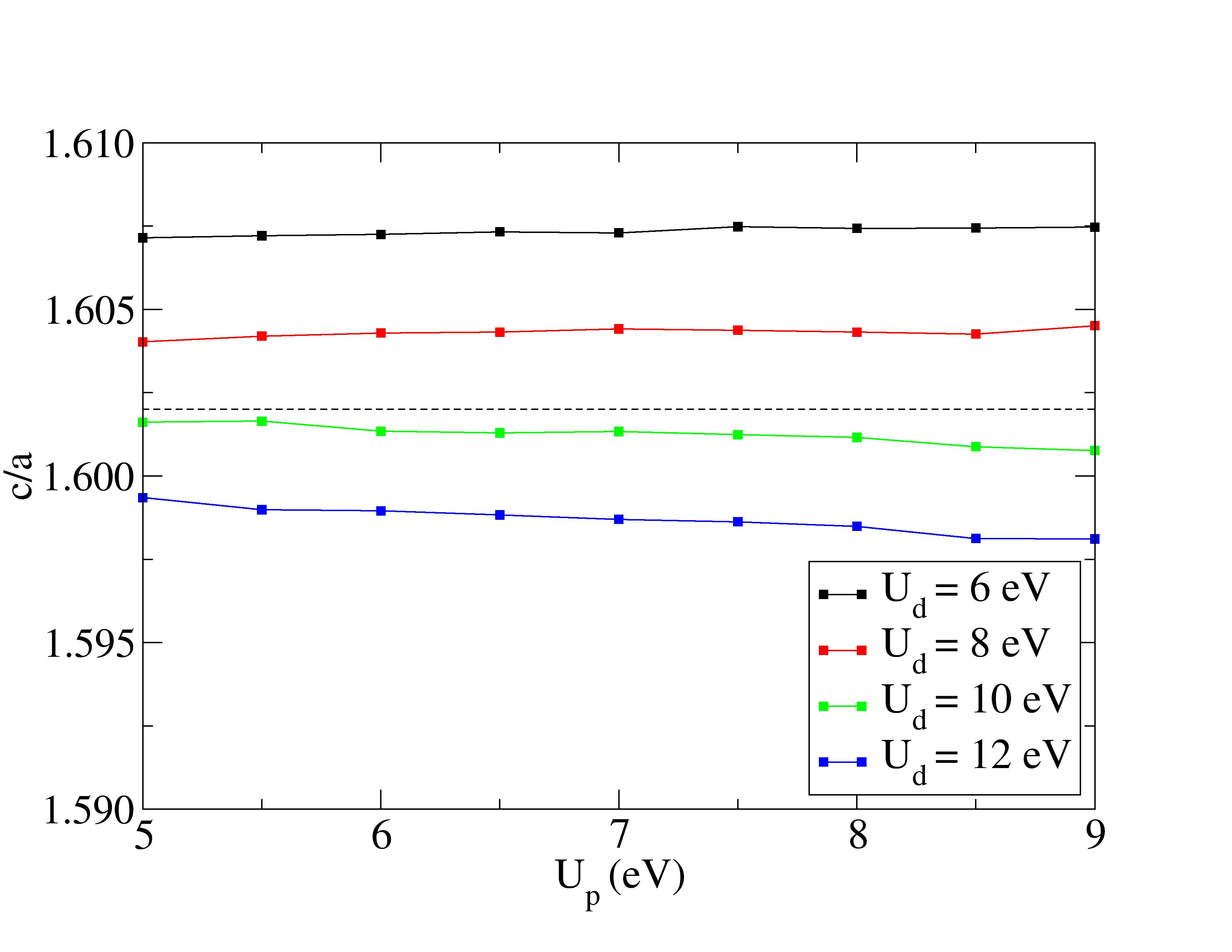 Figure 7
Figure 7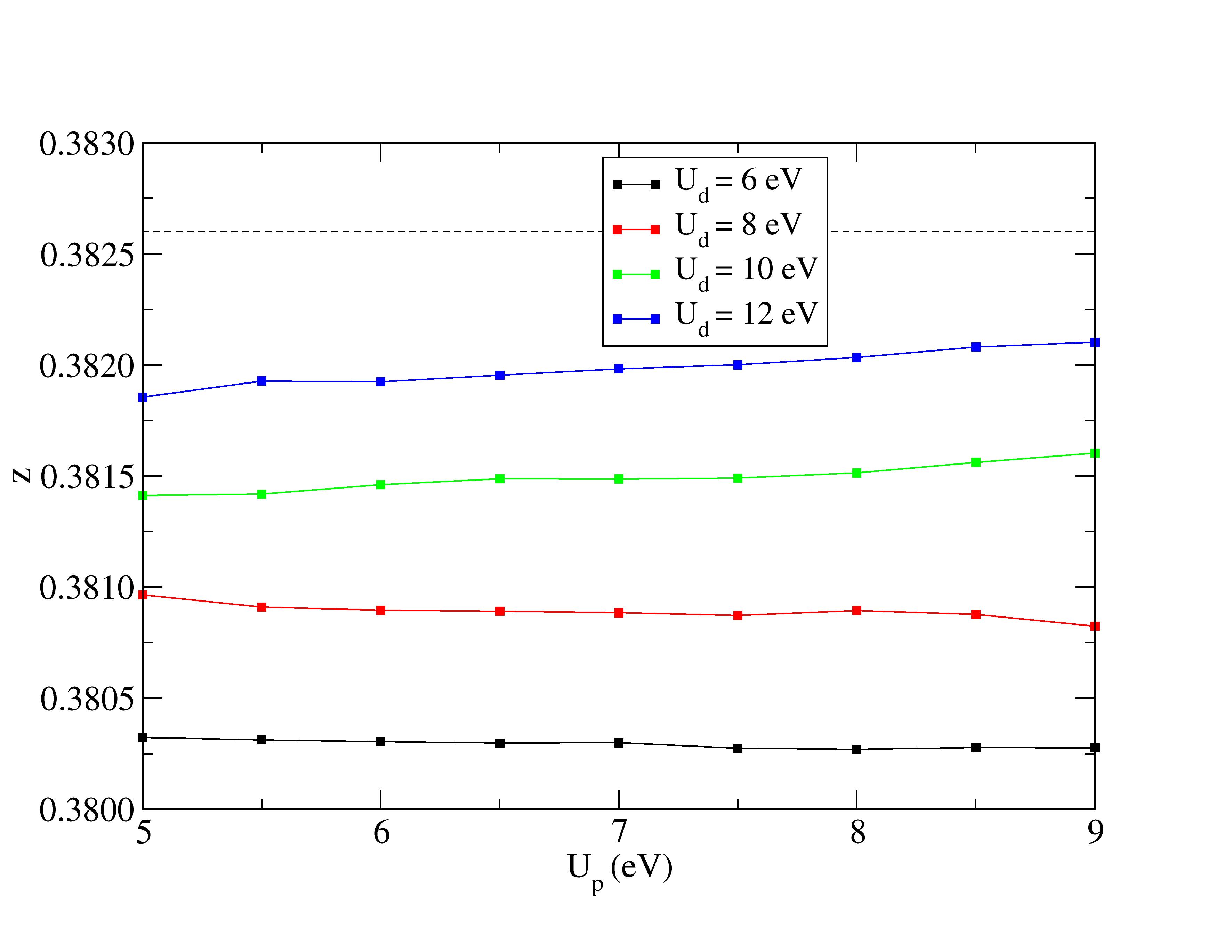 Figure 8
Figure 8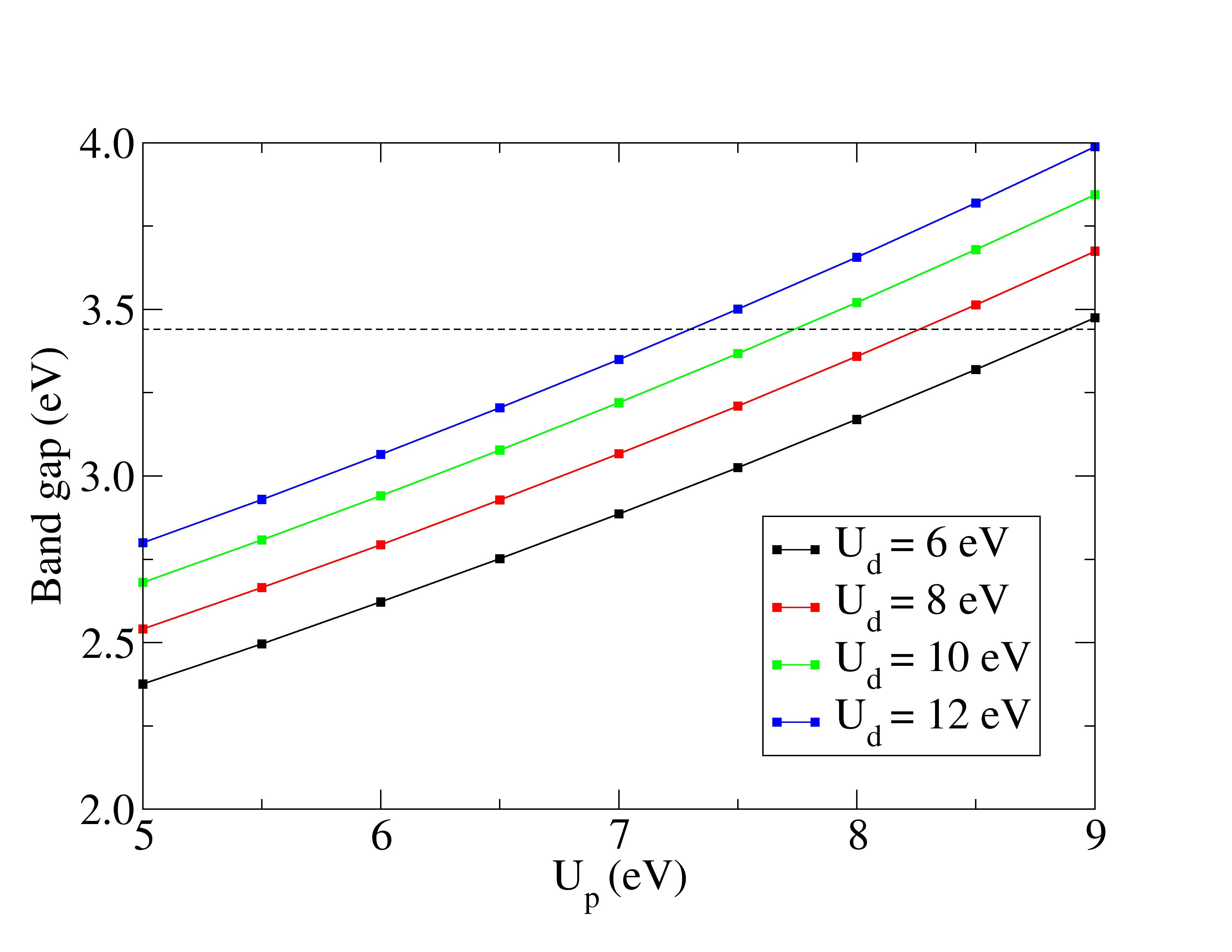 Figure 9
Figure 9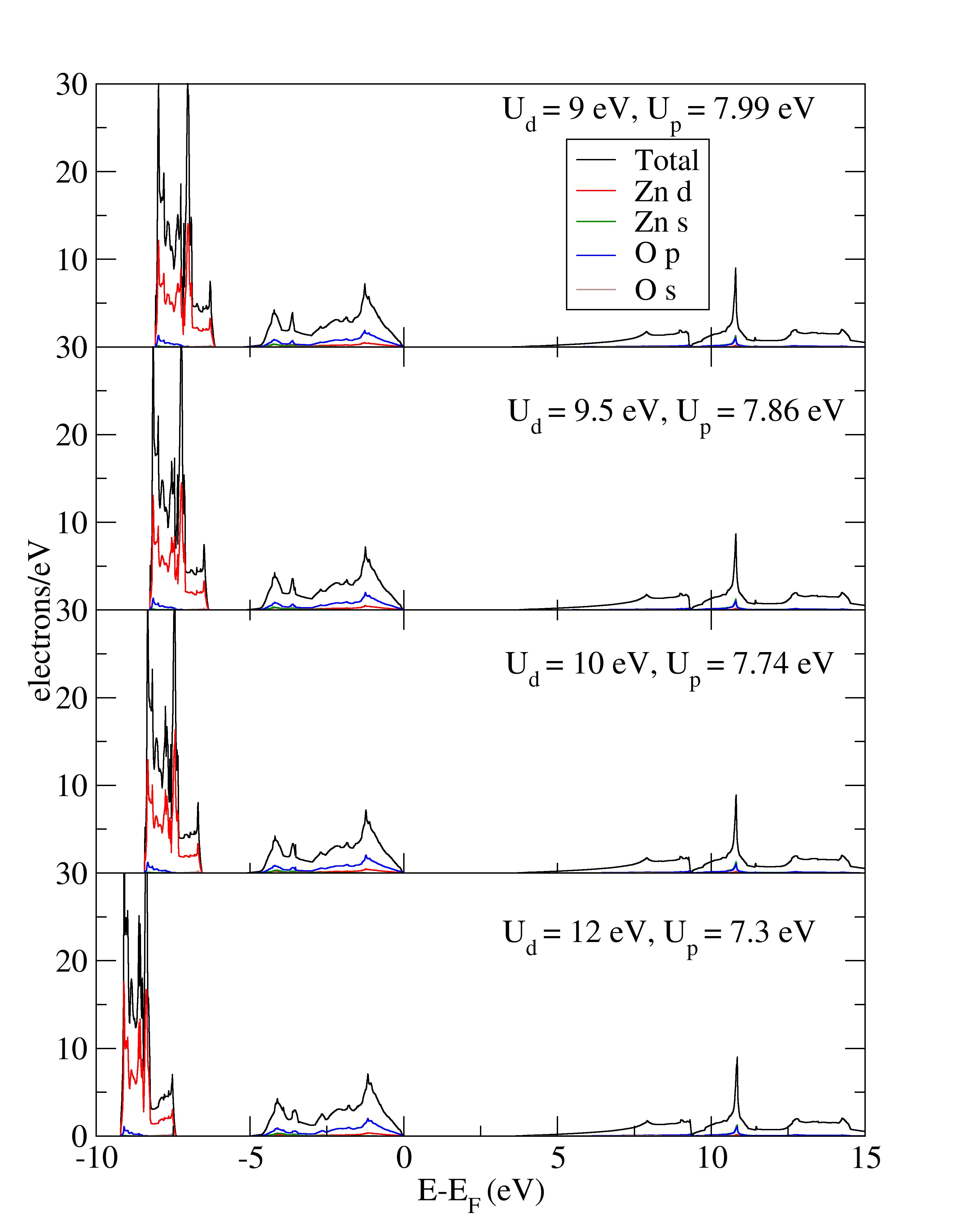 Figure 10
Figure 10| Atom | 2b | (1/3,2/3,0), (2/3,1/3,1/2)\\ O | 2b | (1/3, 2/3, z),(2/3,1/3, z+1/2)\\ |
| Atom | 2b | (1/3,2/3,0), (2/3,1/3,1/2)\\ O | 2b | (1/3, 2/3, z),(2/3,1/3, z+1/2)\\ |
| 3.2427 | 5.1948 | 1.6020 | 0.3826 | 47.3056 | 3.44\\ LDA | 3.1836 | 5.1497 | 1.6175 | 0.3804 | 45.2022 | 0.7965\\ |
| 3.2427 | 5.1948 | 1.6020 | 0.3826 | 47.3056 | 3.44\\ LDA | 3.1836 | 5.1497 | 1.6175 | 0.3804 | 45.2022 | 0.7965\\ |
|
|
|
|
|
|
|
| 7.99 | 3.2427 | 5.1948 | 1.602 | 0.3826 | 7.8 | 2.5 | 1.2 | 5.3 | 9\\ | 7.86 | 3.113 | 4.987 | 1.602 | 0.3813 | 7.685 | 1.931 | 1.208 | 5.102 | 8.242\\ 10.0 | 7.74 | 3.115 | 4.988 | 1.601 | 0.3815 | 7.875 | 1.888 | 1.387 | 5.137 | 8.412\\ 12.0 | 7.30 | 3.124 | 4.995 | 1.600 | 0.3820 | 8.740 | 1.837 | 2.369 | 4.998 | 9.204\\ Experiment |
| 7.99 | 3.2427 | 5.1948 | 1.602 | 0.3826 | 7.8 | 2.5 | 1.2 | 5.3 | 9\\ | 7.86 | 3.113 | 4.987 | 1.602 | 0.3813 | 7.685 | 1.931 | 1.208 | 5.102 | 8.242\\ 10.0 | 7.74 | 3.115 | 4.988 | 1.601 | 0.3815 | 7.875 | 1.888 | 1.387 | 5.137 | 8.412\\ 12.0 | 7.30 | 3.124 | 4.995 | 1.600 | 0.3820 | 8.740 | 1.837 | 2.369 | 4.998 | 9.204\\ Experiment |
| Mode | 92.5 | 112.1 | 98.4\\ B1 | 258.4 | 299.4 | 258.9\\ A1(TO) | 376.5 | 433.5 | 378.3\\ E1(TO) | 415.8 | 466.5 | 412.1\\ E2 | 447.9 | 495.9 | 438.8\\ B1 | 544.7 | 623.4 | 551.7\\ A1(LO) | 513.6 | 638.4 | 573.5\\ E1(LO) | 532.6 | 646.9 | 592.8\\ |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsZnO doping and properties · Ga2O3 and related materials · Copper-based nanomaterials and applications
Effects of Hubbard term correction on the structural parameters and
electronic properties of wurtzite ZnO
E. S. Goh
School of Physics, Universiti Sains Malaysia, 11800 USM Penang, Malaysia
J. W. Mah
School of Physics, Universiti Sains Malaysia, 11800 Penang, Malaysia
T. L. Yoon
School of Physics, Universiti Sains Malaysia, 11800 Penang, Malaysia
Abstract
The effects of including the Hubbard on-site Coulombic correction to the structural parameters and valence energy states of wurtzite ZnO was explored. Due to the changes in the structural parameters caused by correction of hybridization between Zn d states and O p states, suitable parameters of Hubbard terms have to be determined for an accurate prediction of ZnO properties. Using the LDA+ method by applying Hubbard corrections to Zn 3d states and to O 2p states, the lattice constants were underestimated for all tested Hubbard parameters. The combination of both and correction terms managed to widen the band gap of wurtzite ZnO to the experimental value. Pairs of and parameters with the correct positioning of d-band and accurate bandwidths were selected, in addition to predicting an accurate band gap value. Inspection of vibrational properties, however, revealed mismatches between the estimated gamma phonon frequencies and experimental values. The selection of Hubbard terms based on electronic band properties alone cannot ensure an accurate vibrational description in LDA+ calculation.
keywords:
density functional theory, Hubbard correction, wurtzite ZnO, electronic
1 Introduction
Zinc oxide had been known as a relatively low cost and readily synthesized material. It has the properties of a polar semiconductor with a wide band gap of 3.44 eV with potential applications in the optoelectronic industries [1]. Numerous theoretical calculation on the properties of ZnO had been carried out using density functional theory (DFT). However, it is widely known that a standard DFT calculation typically suffers from a band gap problem, where the band gap of a material is grossly underestimated [2]. This could lead to an inaccurate estimation of the electronic properties of a material such as ZnO, which is a potential material for the optoelectronic industry.
The band gap problem of DFT calculation can be addressed by including the GW approximation in which the self-energy of a many-body system of electrons is taken into account [3]. Zhang et al. estimated that the band gap of wurtzite ZnO to be within the range of 2.82 eV to 4.54 eV by using various types of GW approximations [4]. An alternative way to improve the band gap is to employ the hybrid exchange correlation functionals in a DFT calculation. Using the HSE06 functional, Zhou predicted the band gap of wurtzite ZnO to be 2.79 eV [5]. While both GW approximation and hybrid functional have proved to be effective in mitigating the band gap in a standard DFT calculation, a major downside is the requirement of a computational power much higher than that required by a standard DFT computation. This has limited the feasibility of conducting a realistic prediction of properties of a material via DFT.
DFT+U calculation has emerged as a means to improve the electronic properties prediction at a computational cost comparable to that required by a standard DFT calculation. In a DFT+U calculation, Hubbard-type interactions are included in the standard exchange correlation functional of local density approximation (LDA) or generalized-gradient approximation (GGA) through the Hubbard parameters U and J [6, 7]. A method of estimating the values for the Hubbard parameters U and J had been provided by Cococcioni et al. through the linear response approach [8]. However, the linear response approach, which has been widely used for the open shell system, is less than ideal in the case of a closed-shell system such as ZnO with a full electronic shell of d orbitals. Numerical reliability is an issue of concern in the case of completely full localized bands which exhibit a very small response to linear perturbation [9].
In this work, the dependence of structural parameters and electronic properties of wurtzite ZnO on the different values of Hubbard parameters U and J is investigated. A series of DFT+U calculation is carried out with different values of both Hubbard parameters associated with Zn and O. While the Hubbard parameters are introduced to improve electronic properties, the structural parameters of a material are modified alongside with the electronic properties; the selection of suitable values of Hubbard parameters depends on the both structural and electronic properties. A similar work on the LDA+U and GGA+U calculation of wurtzite ZnO had been carried out by Huang et al. [10] by using the electronic calculation package VASP. However, different results are obtained in this work through the use of different pseudopotentials.
2 Computational Methods
The experimental structural parameters of wurtzite ZnO at a=3.2427 Å and c= 5.1948 Å with wurtzite parameter z=0.3826 as discovered by Sabine and Hogg [11] using X-ray crystallography method is used as the reference structural data in this work. A standard DFT geometrical optimization is first performed on the primitive wurtzite ZnO unit cell, followed by a self-consistent DFT calculation to study its electronic properties. Hubbard term is then added to the d-orbitals of the Zn atoms, ranging from 2 eV to 14 eV in the interval of 2 eV. The second part of this research studies the effect of including both Hubbard term to Zn d-orbitals and to O p-orbitals, where the values ranges from 5 eV to 9 eV. The changes to lattice constants and wurtzite parameter z as well as the band gap and valence band width are investigated.
All calculations in this work are completed using the ABINIT electronic package [12] within the framework of projector-augmented-wave (PAW) potentials [13] and LDA exchange correlation functionals. The PAW potentials used are from the datasets provided by Jollet et al. [14]. The plane wave basis sets are expanded to kinetic energy cutoff of 34 Hartree whereas the Monkhorst-Pack k-point mesh is set to an array of at gamma centred grid.
The LDA+U calculations are performed using full localized limit (FLL) double-counting correction [6]. The double-counting correction is necessary in a LDA+U calculation to avoid double counting of correlation part in localized electrons. The Hubbard term J is set to zero for all calculations; the rotationally invariant LDA+U form proposed by Dudarev et al. [7] is equivalent to the FLL method with J=0 and U in place of [15].
3 Results and Discussion
3.1 Standard DFT result
Wurtzite ZnO possesses a space group number of 186 with hexagonal symmetry and the following Wyckoff positions (see Table 1).