TL;DR
This paper investigates the pressure-induced metal-insulator transition in a two-dimensional stacked hydrogen molecule system, revealing a Mott-Hubbard-type transition with abrupt changes in electronic and structural properties.
Contribution
It introduces a combined exact diagonalization and ab initio approach to model the Mott-Hubbard transition in 2D hydrogen layers, including wave function readjustment and long-range interactions.
Findings
Discontinuous transition from molecular insulator to quasiatomic metal.
No intermediate quasiatomic insulating phase observed.
Abrupt changes in bond length and electronic properties at transition.
Abstract
We analyze the pressure-induced metal-insulator transition in a two-dimensional vertical stack of molecules in x-y plane, and show that it represents a striking example of the Mott-Hubbard-type transition. Our combined exact diagonalization approach, formulated and solved in the second quantization formalism, includes also simultaneous ab initio readjustment of the single-particle wave functions, contained in the model microscopic parameters. The system is studied as a function of applied side force (generalized pressure), both in the -molecular and -quasiatomic states. Extended Hubbard model is taken at the start, together with longer-range electron-electron interactions incorporated into the scheme. The stacked molecular plane transforms discontinuously into a (quasi)atomic state under the applied force via a two-step transition: the first between molecular insulating…
Click any figure to enlarge with its caption.
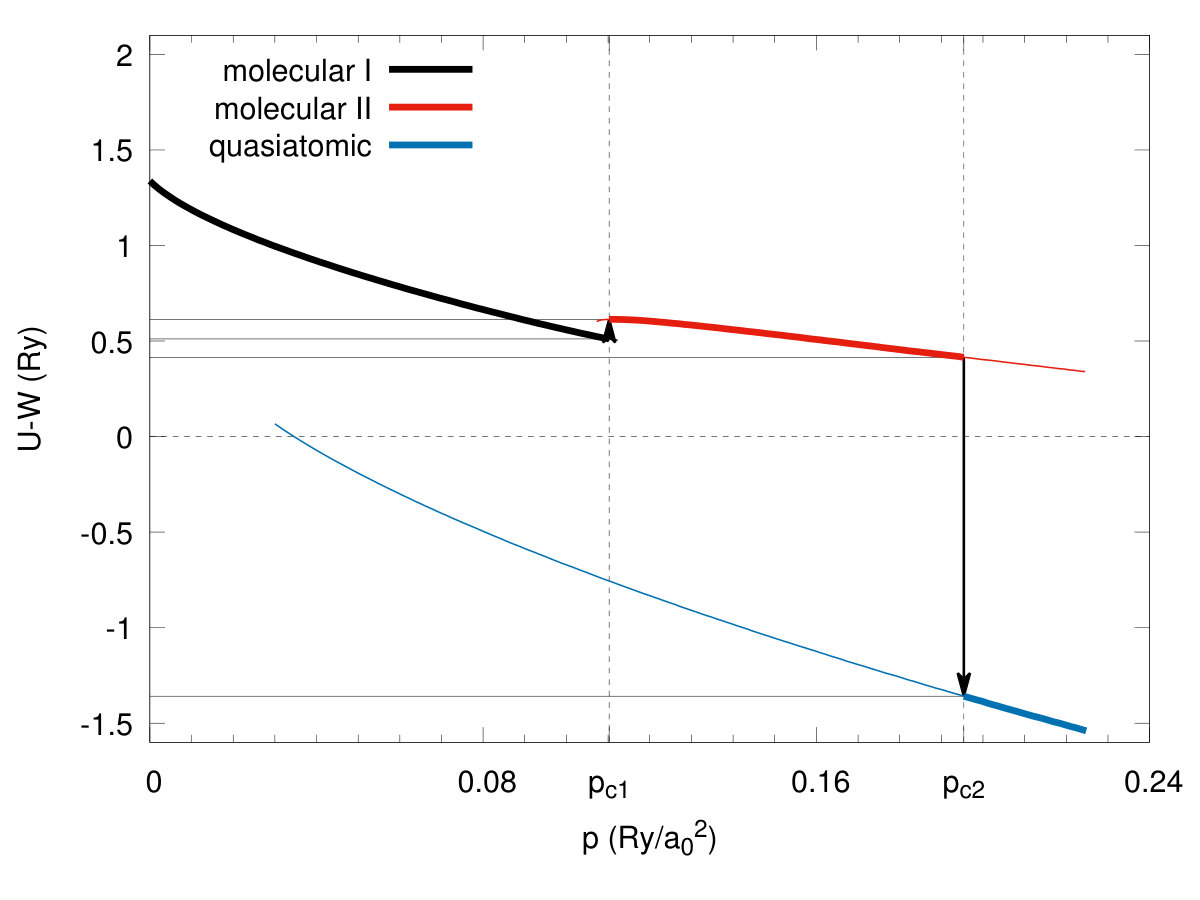 Figure 1
Figure 1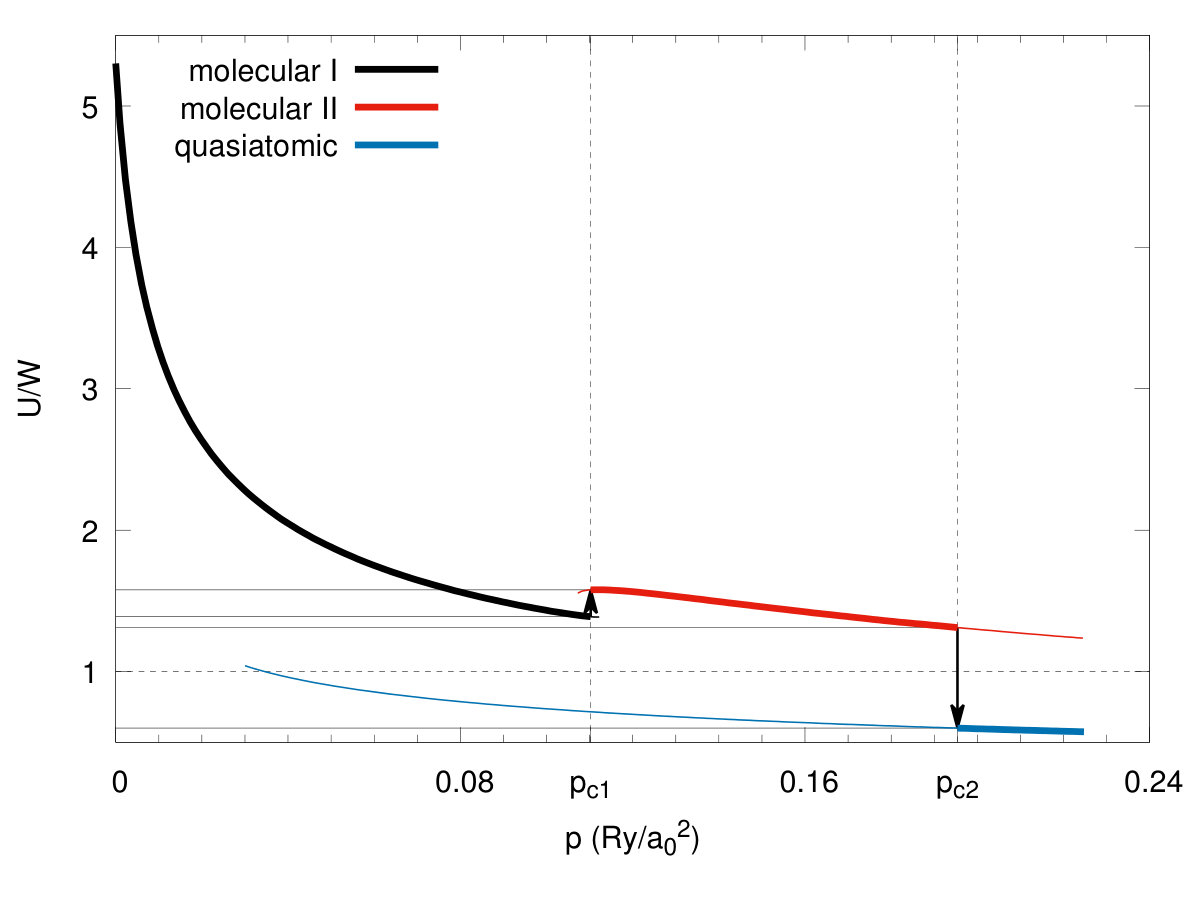 Figure 2
Figure 2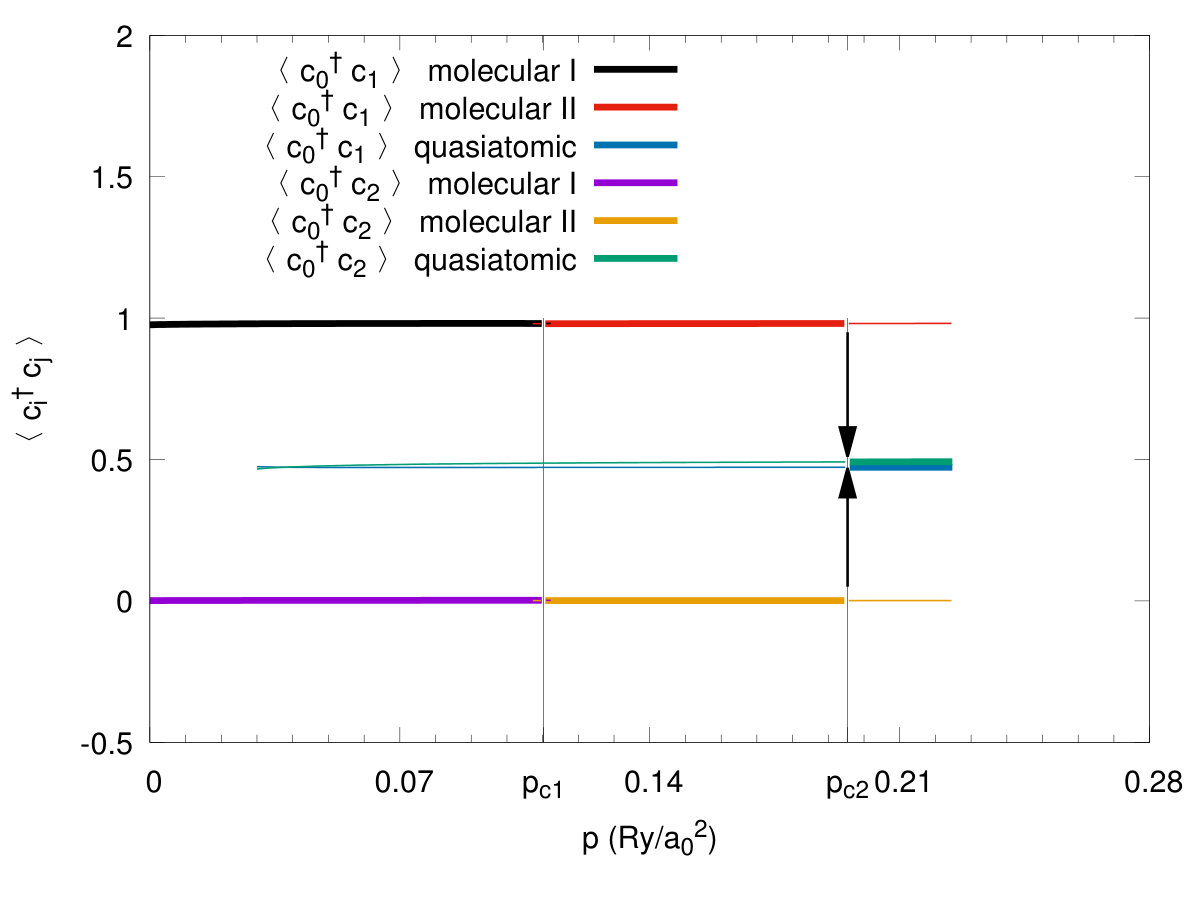 Figure 3
Figure 3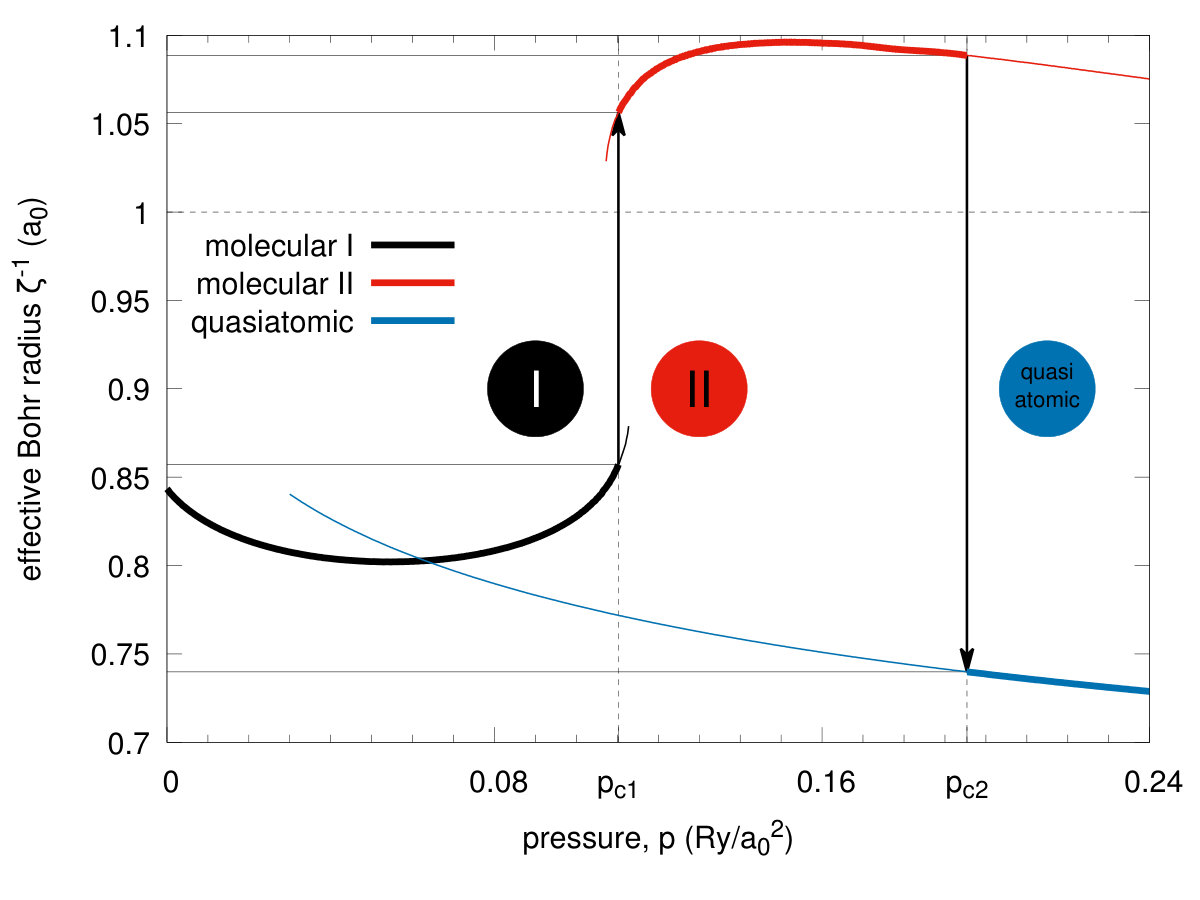 Figure 4
Figure 4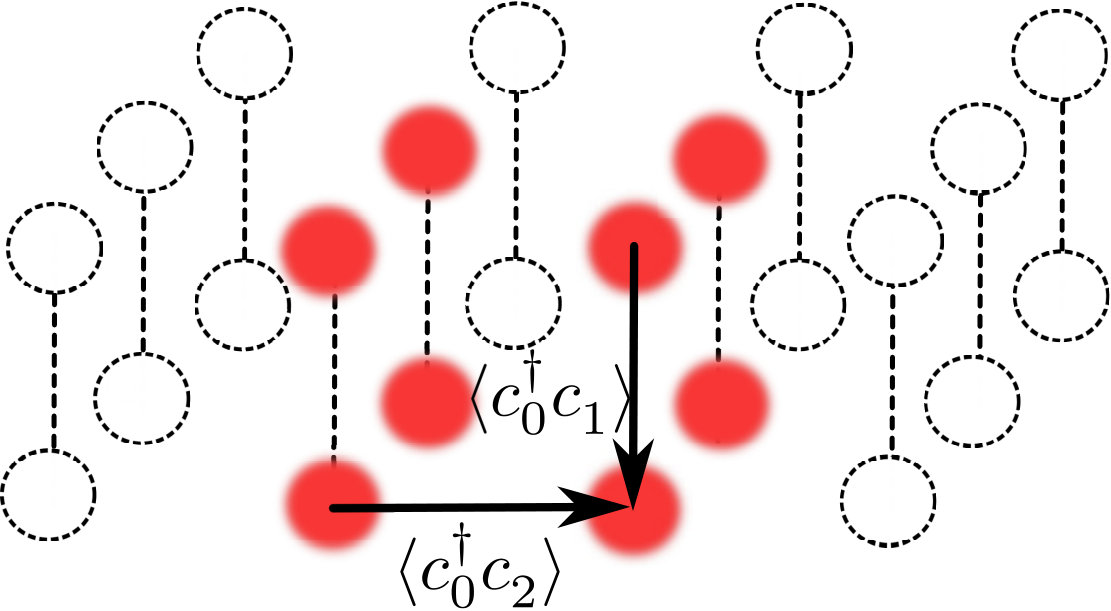 Figure 5
Figure 5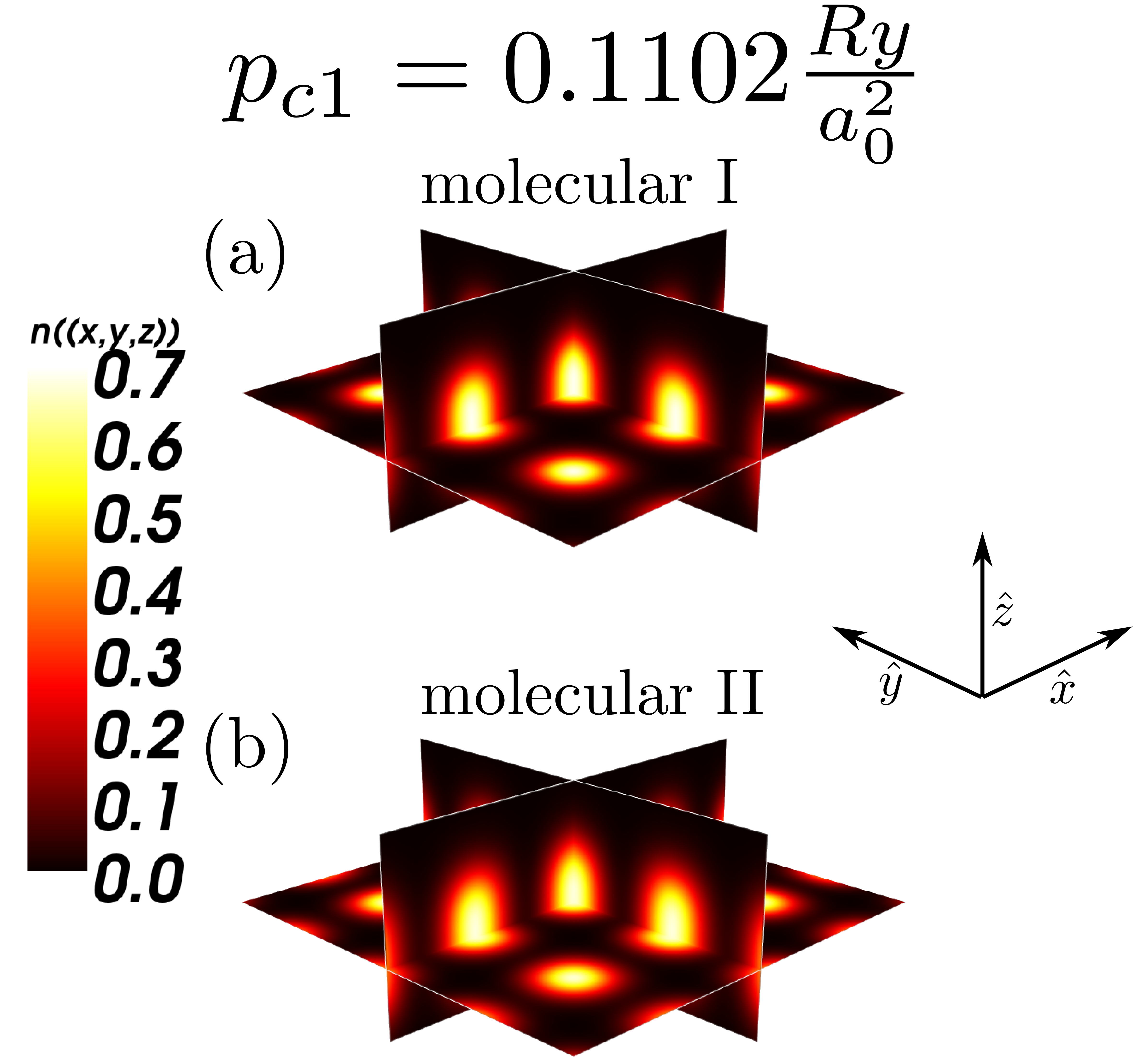 Figure 6
Figure 6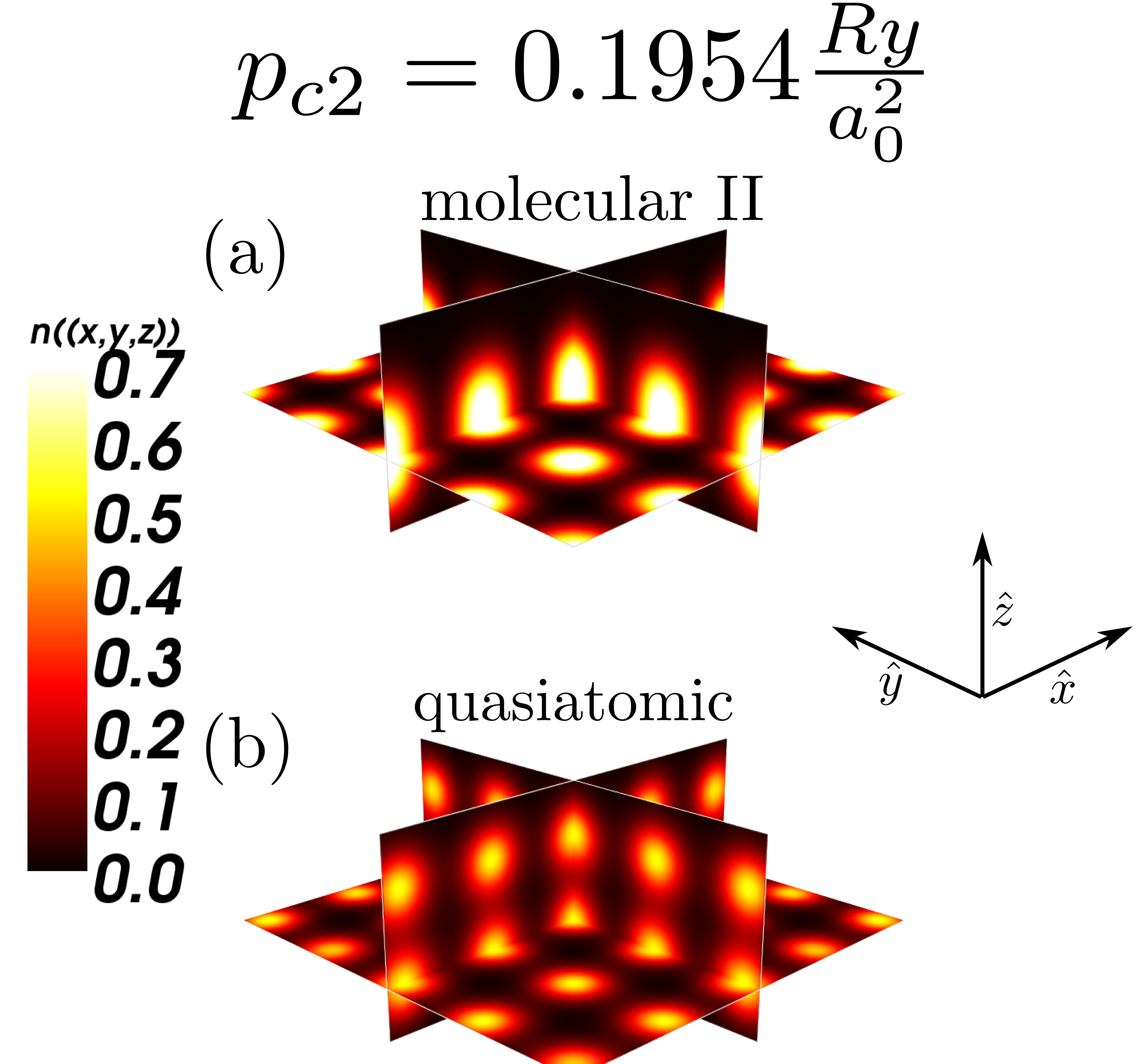 Figure 7
Figure 7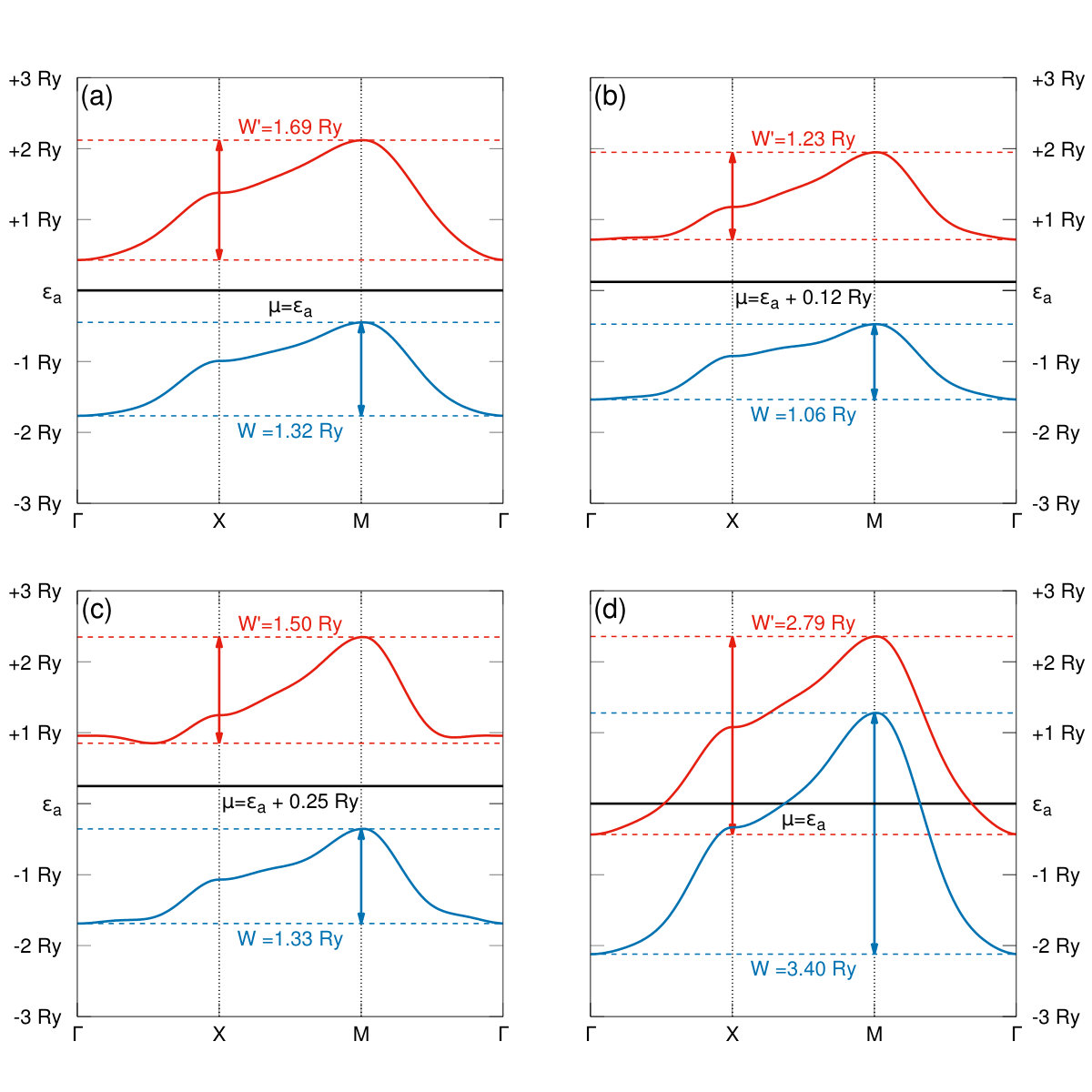 Figure 8
Figure 8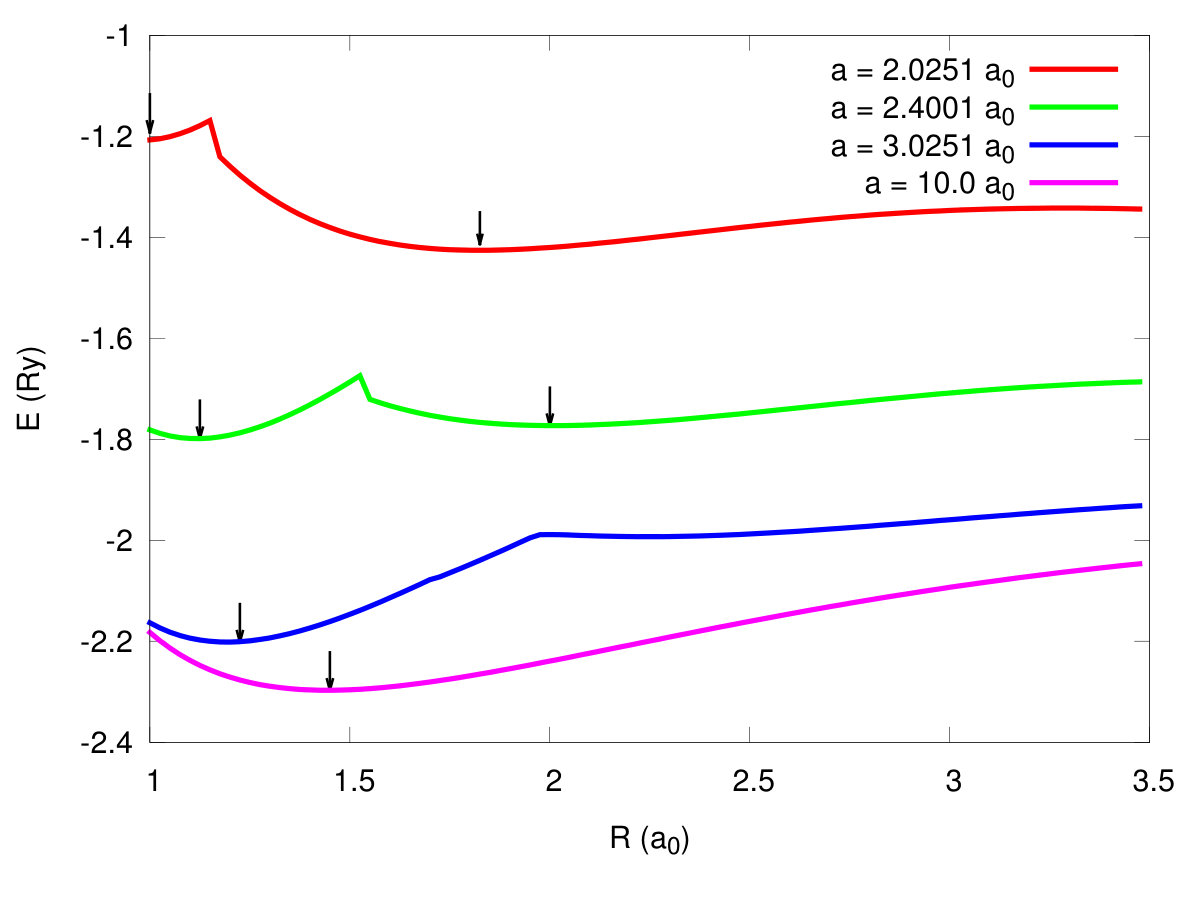 Figure 9
Figure 9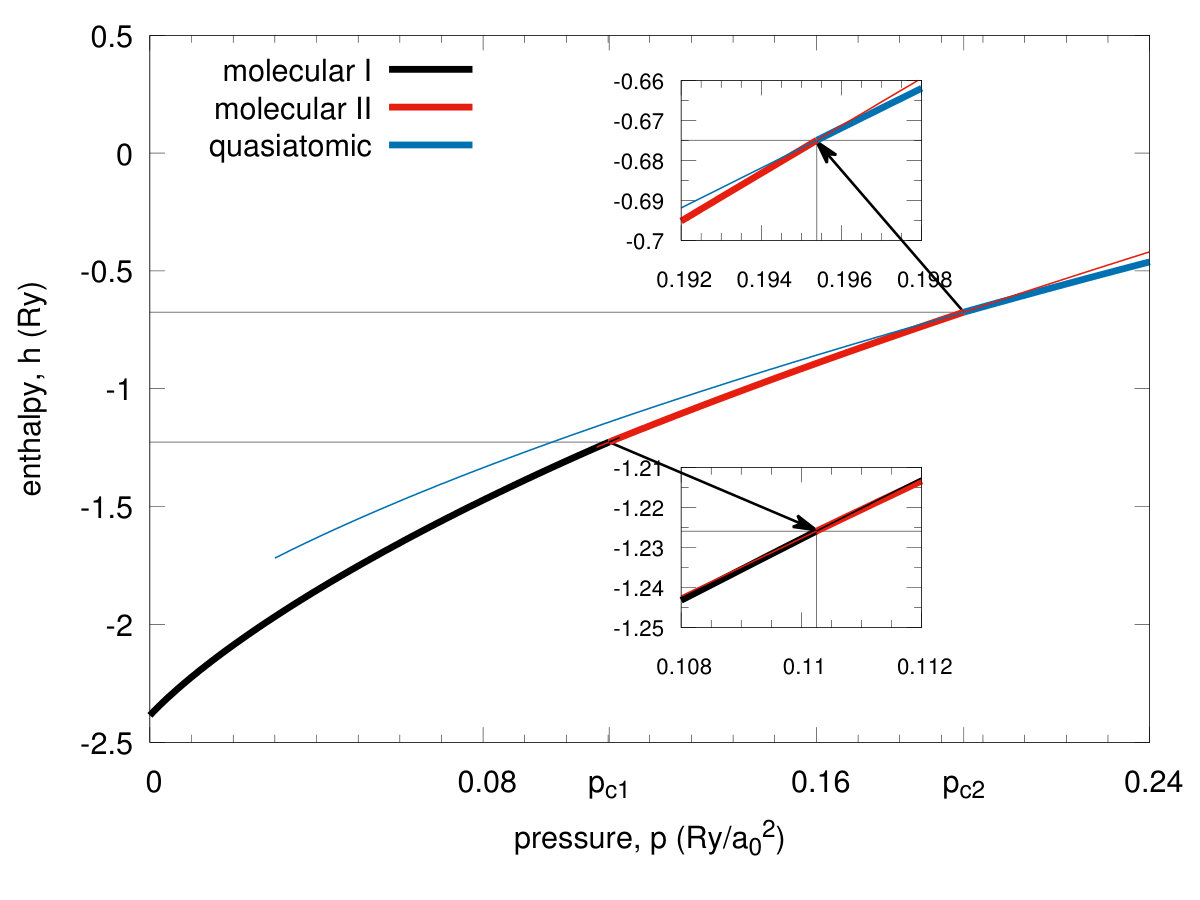 Figure 10
Figure 10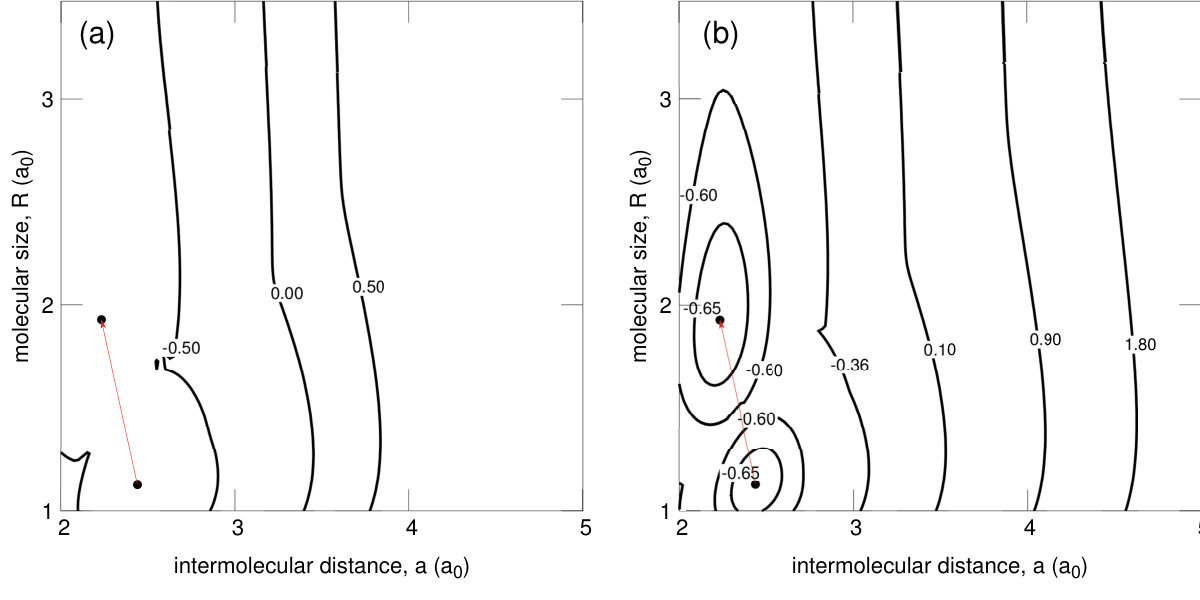 Figure 11
Figure 11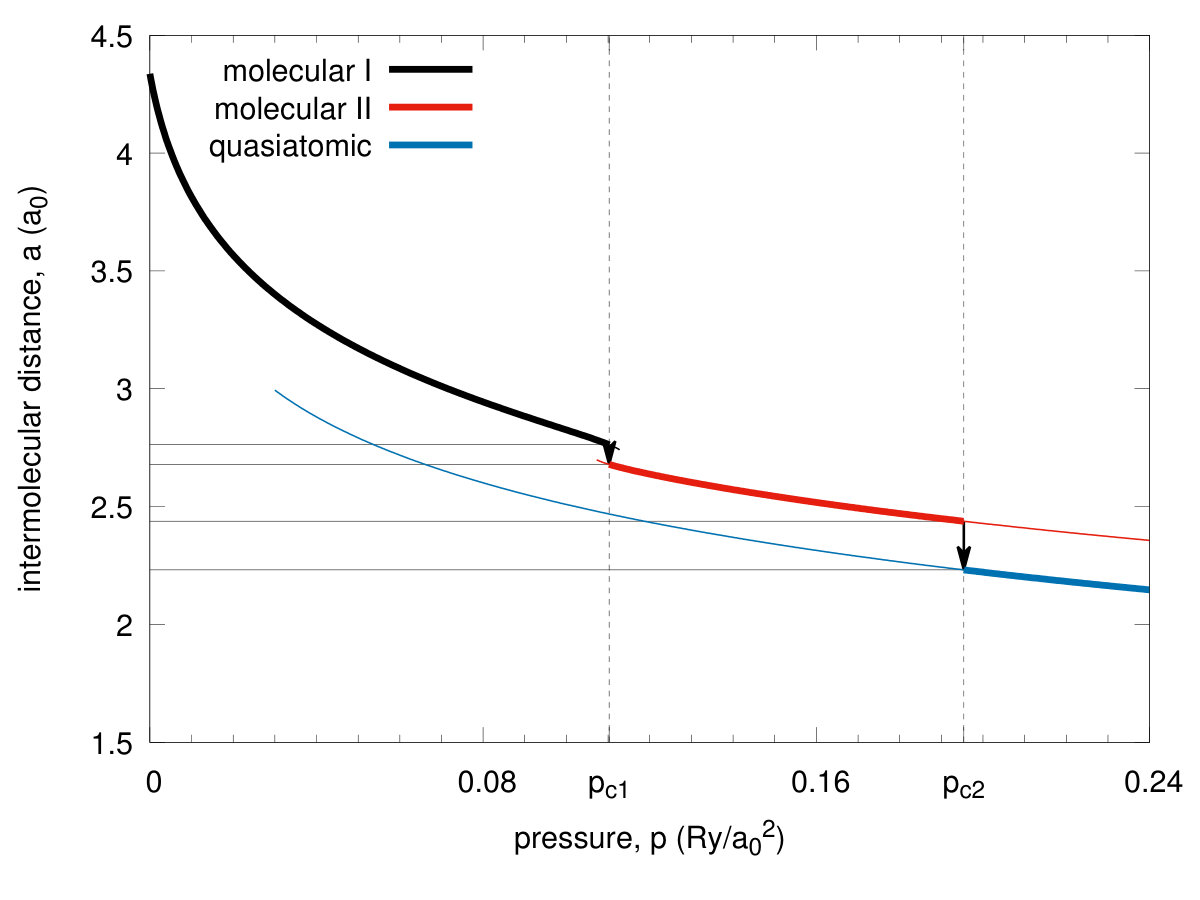 Figure 12
Figure 12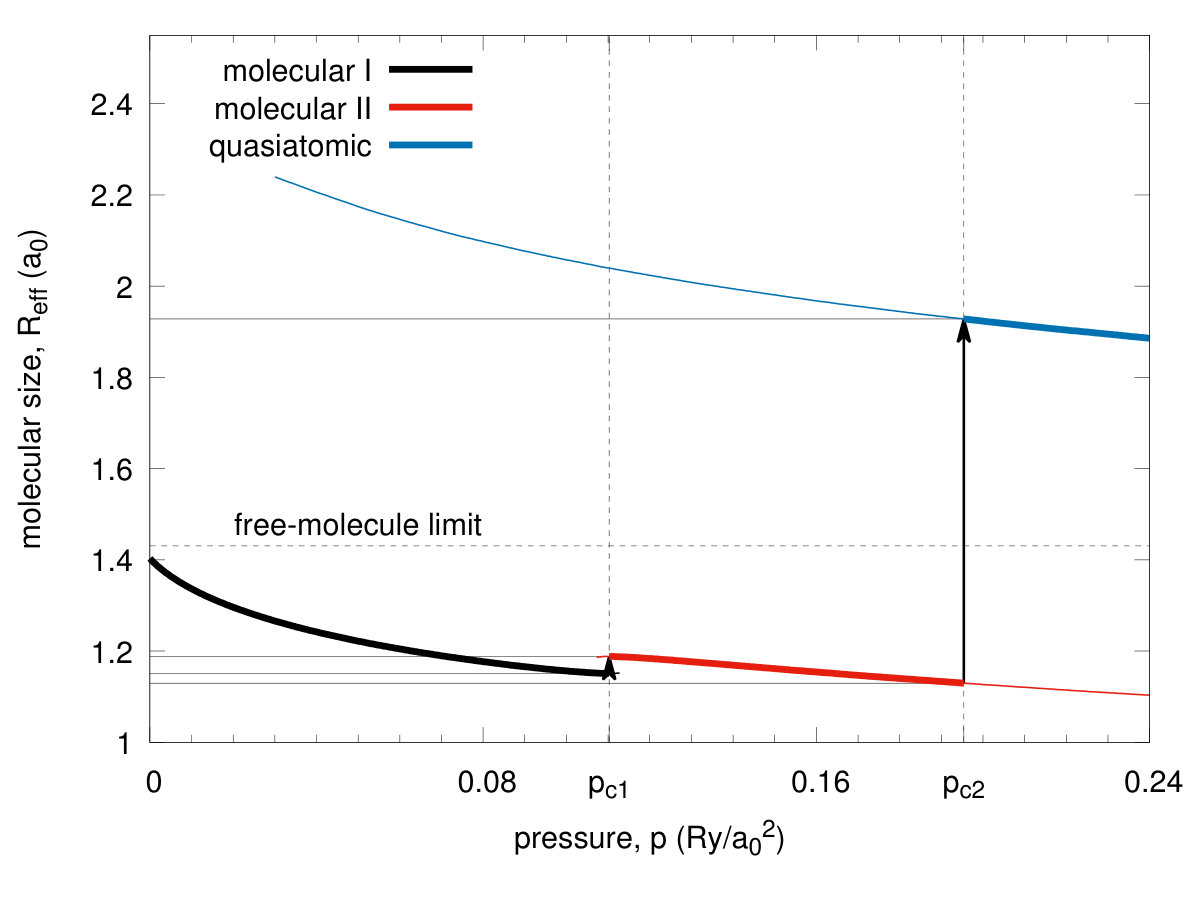 Figure 13
Figure 13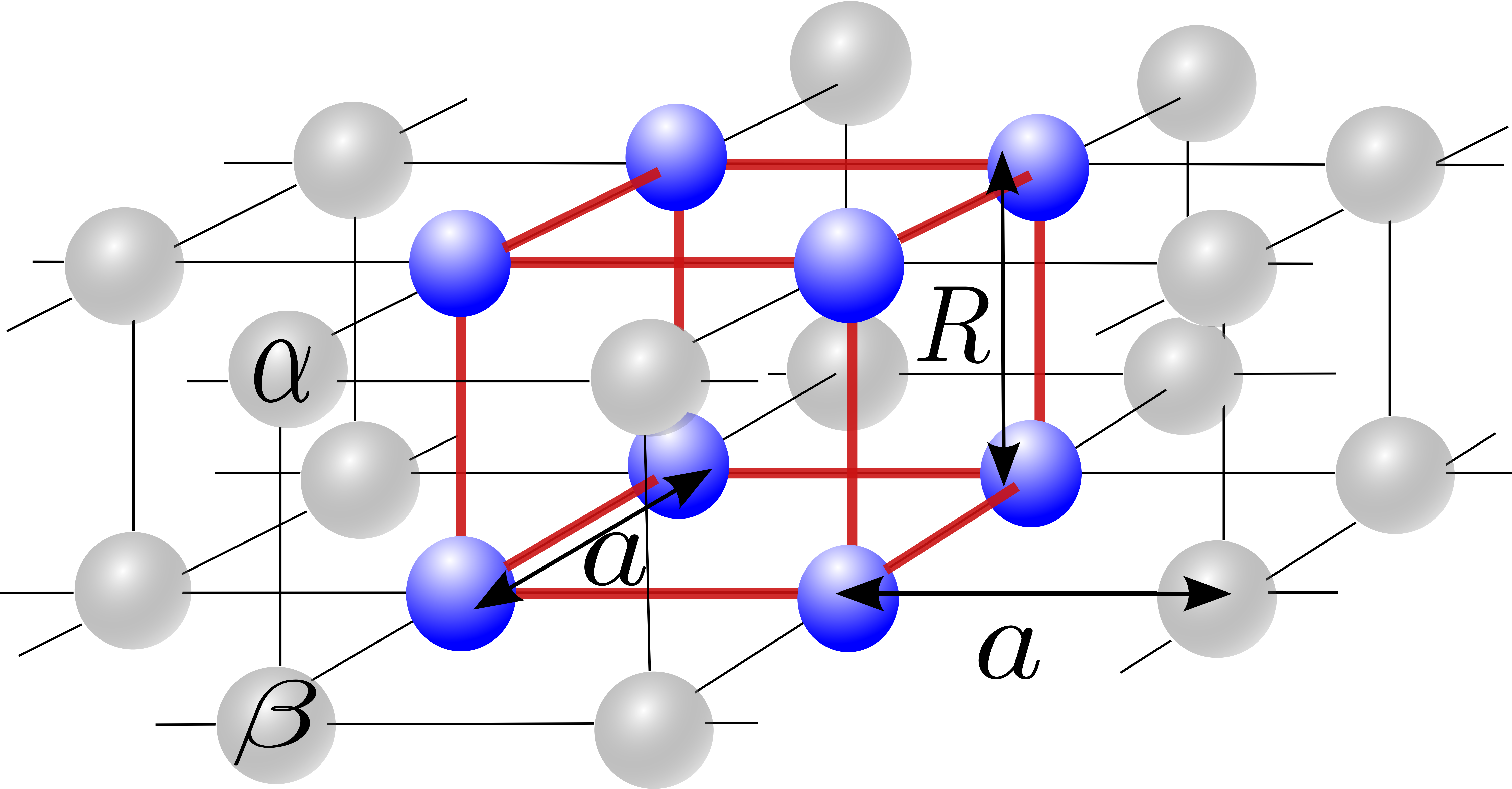 Figure 14
Figure 14 Figure 15
Figure 15 Figure 16
Figure 16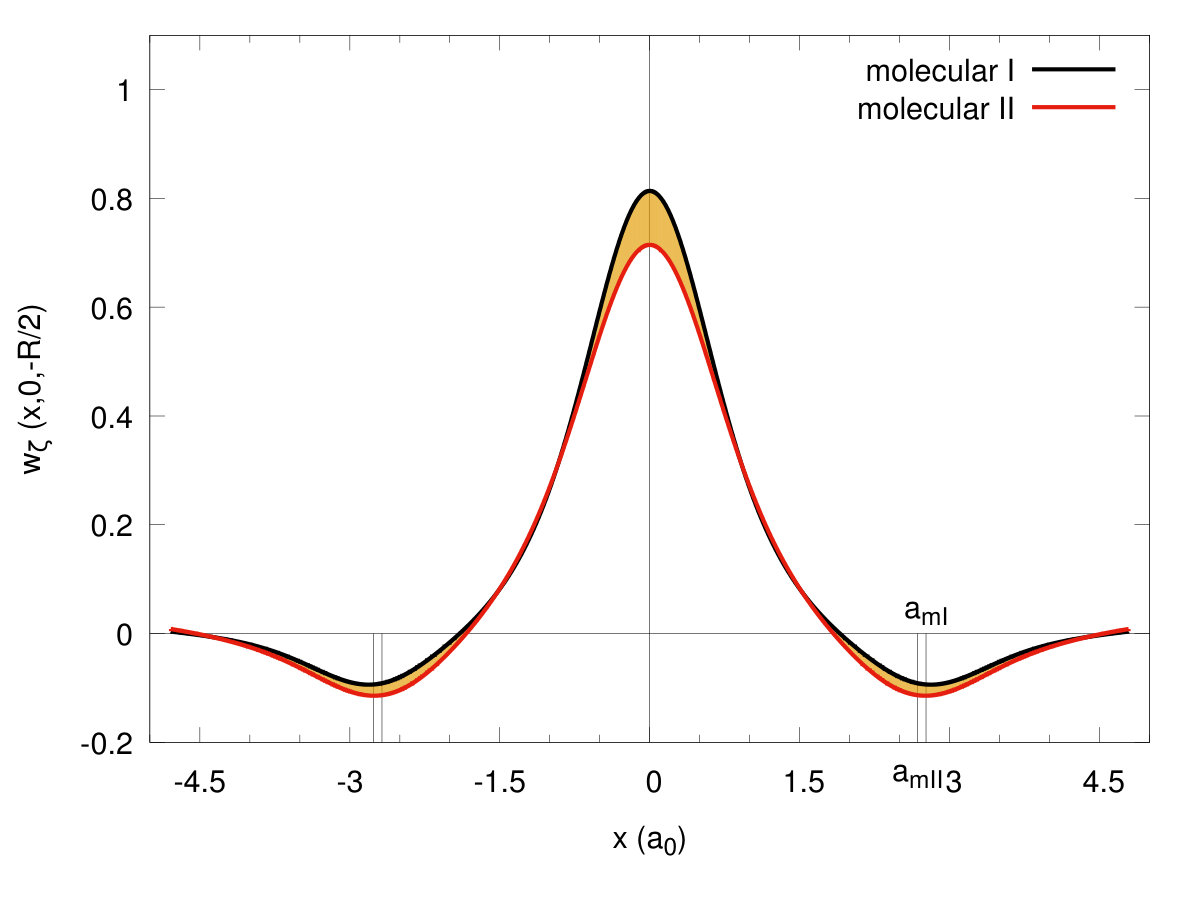 Figure 17
Figure 17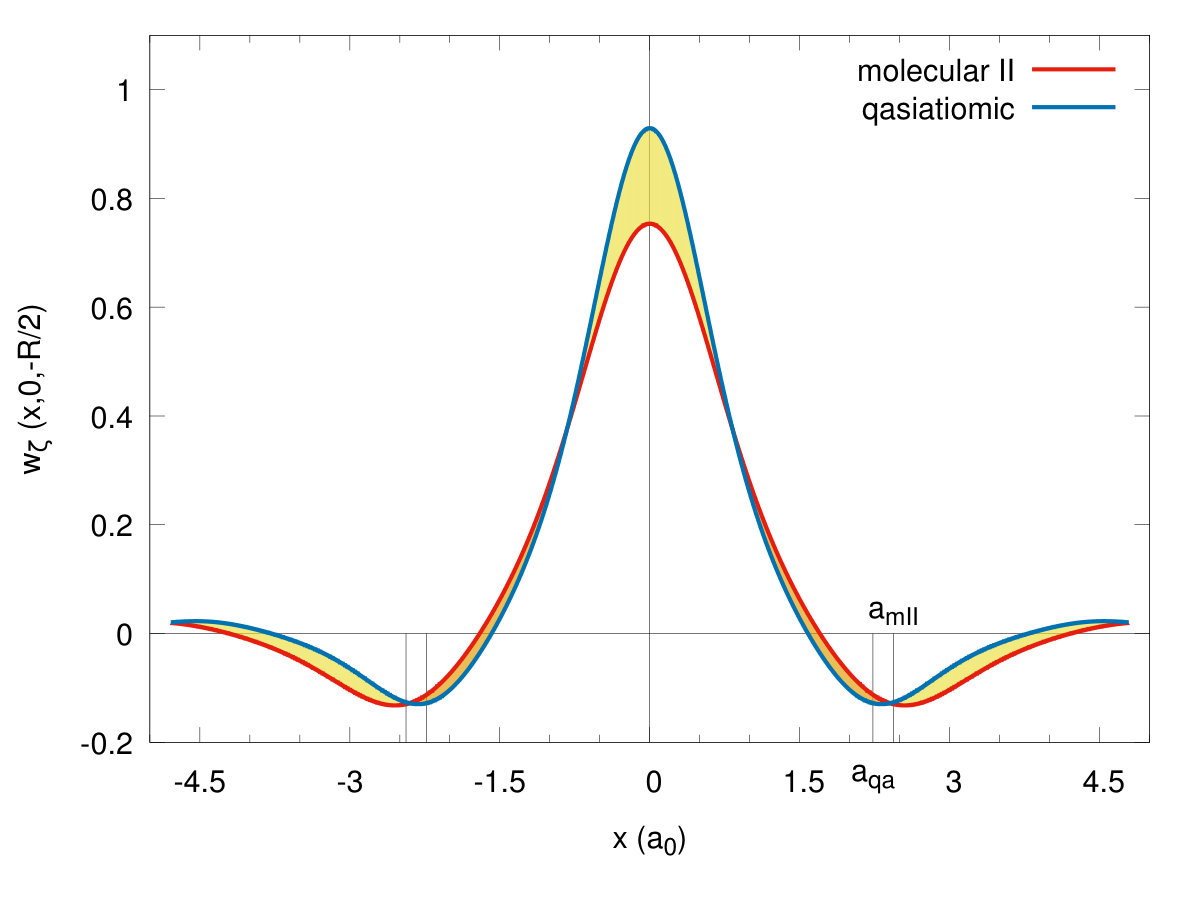 Figure 18
Figure 18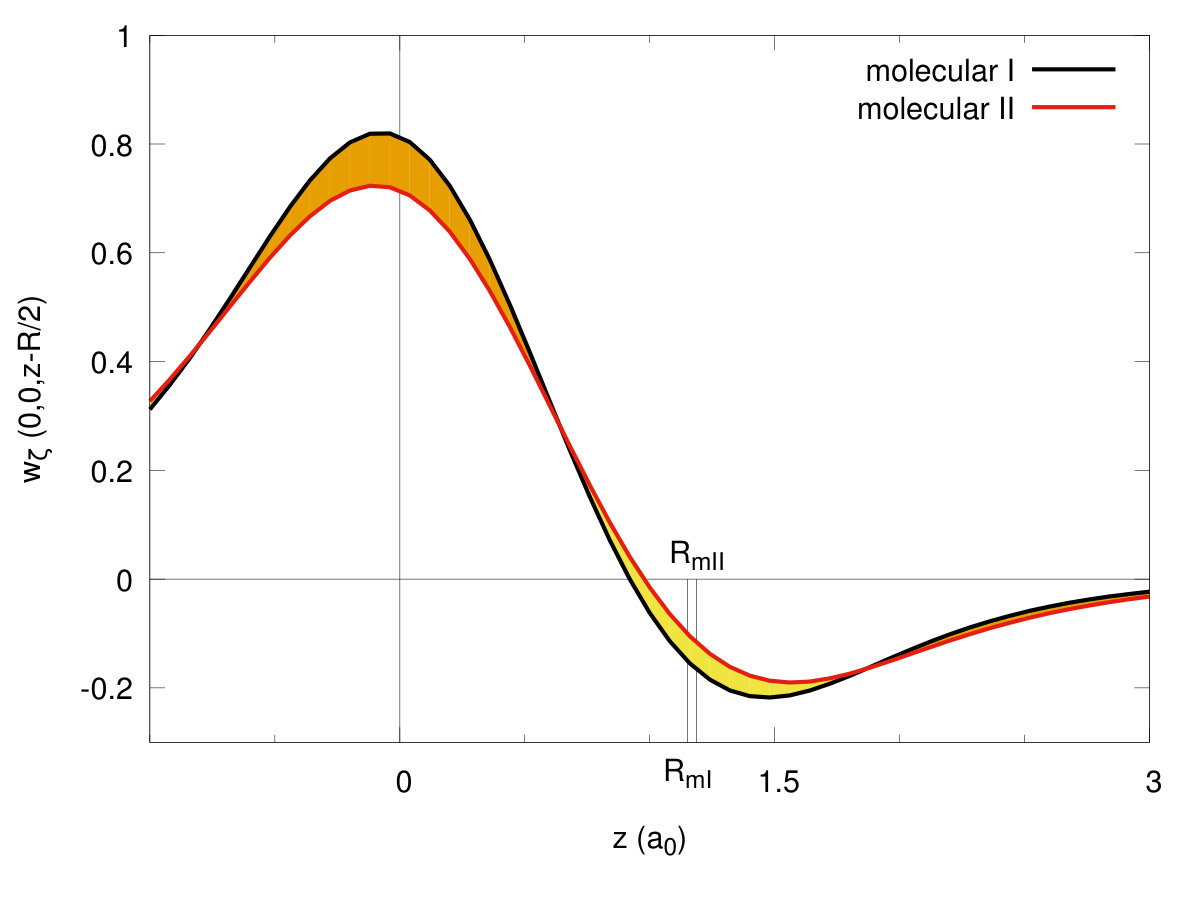 Figure 19
Figure 19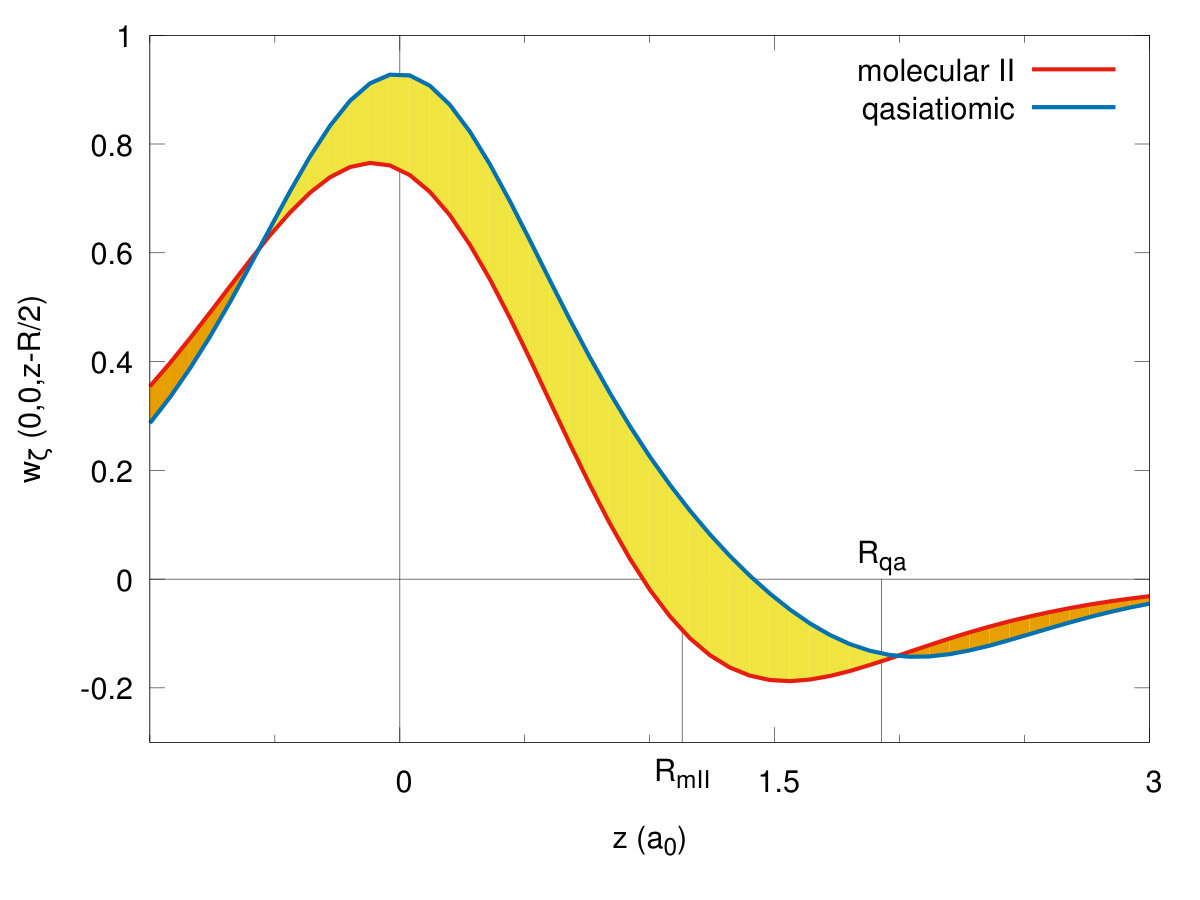 Figure 20
Figure 20| molecular I | 2.7626 | 1.1511 | 1.1667 | 1.8268 | 1.0725 | 0.7173 | -1.1985 | -0.1933 | |
|---|---|---|---|---|---|---|---|---|---|
| molecular II | 2.6791 | 1.1881 | 0.9466 | 1.6751 | 0.9847 | 0.7289 | -1.1177 | -0.1422 | |
| molecular II | 2.4378 | 1.1296 | 0.9186 | 1.7486 | 1.0244 | 0.7945 | -1.2456 | -0.1596 | |
| quasiatomic | 2.2313 | 1.9281 | 1.3516 | 2.0392 | 0.9380 | 0.8760 | -0.7660 | -0.3884 |
| molecular I | 2.7626 | 1.1511 | 0.8571 | 1.3870 | 0.3103 | |
|---|---|---|---|---|---|---|
| molecular II | 2.6791 | 1.1881 | 1.0564 | 1.5778 | 0.3943 | |
| molecular II | 2.4378 | 1.1296 | 1.0887 | 1.3112 | 0.4466 | |
| quasiatomic | 2.2313 | 1.9281 | 0.7398 | 0.6000 | 0.3316 |
| phase | direction of the mode | |||||
| molecular I | 4.3371 | 1.4031 | -2.3858 | or | ||
| or | ||||||
| 0.01837 | ||||||
| 0.08263 Ry | ||||||
| molecular I | 2.7626 | 1.1511 | -2.0674 | and | ||
| and | ||||||
| 0.00452 | ||||||
| 0.1261 Ry | ||||||
| molecular II | 2.6791 | 1.1881 | -2.0173 | and | ||
| and | ||||||
| 0.00557 | ||||||
| 0.13137 Ry | ||||||
| molecular II | 2.4378 | 1.1296 | -1.8362 | and | ||
| and | ||||||
| 0.00402 | ||||||
| 0.14762 Ry | ||||||
| quasiatomic | 2.2313 | 1.9281 | -1.6478 | and | ||
| and | ||||||
| 0.00162 | ||||||
| 0.14104 Ry | ||||||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Code & Models
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Metallization of solid molecular hydrogen in two dimensions:
Mott-Hubbard-type transition
Andrzej Biborski
Academic Centre for Materials and Nanotechnology, AGH University of Science and Technology, al. Mickiewicza 30, PL-30-059 Kraków, Poland
Andrzej P. Kądzielawa
Marian Smoluchowski Institute of Physics, Jagiellonian University, ulica Łojasiewicza 11, PL-30-348 Kraków, Poland
Józef Spałek
Marian Smoluchowski Institute of Physics, Jagiellonian University, ulica Łojasiewicza 11, PL-30-348 Kraków, Poland
Abstract
We analyze the pressure-induced metal-insulator transition in a two-dimensional vertical stack of molecules in (x-y) plane, and show that it represents a striking example of the Mott-Hubbard-type transition. Our combined exact diagonalization approach, formulated and solved in the second quantization formalism, includes also simultaneous ab initio readjustment of the single-particle wave functions, contained in the model microscopic parameters. The system is studied as a function of applied side force (generalized pressure), both in the -molecular and -quasiatomic states. Extended Hubbard model is taken at the start, together with longer-range electron-electron interactions incorporated into the scheme. The stacked molecular plane transforms discontinuously into a (quasi)atomic state under the applied force via a two-step transition: the first between molecular insulating phases and the second from the molecular to the quasiatomic metallic phase. No quasiatomic insulating phase occurs. All the transitions are accompanied by an abrupt changes of the bond length and the intermolecular distance (lattice parameter), as well as by discontinuous changes of the principal electronic properties, which are characteristic of the Mott–Hubbard transition here associated with the jumps of the predetermined equilibrium lattice parameter and the effective bond length. The phase transition can be interpreted in terms of the solid hydrogen metallization under pressure exerted by e.g., the substrate covered with a monomolecular film of the vertically stacked molecules. Both the Mott and Hubbard criteria at the insulator to metal transition are discussed.
pacs:
71.30.+h 71.27.+a 71.15.-m 31.15.A-,
I Motivation
Hydrogen is the first and the simplest of elements in the Periodic Table, with an elementary structure of the energy levels. Also, the molecule represents the testing ground of quantum-mechanical methods Kolos; Pachucki. This elementary nature of the atomic or molecular energy levels transforms into an involved manifold of states and available energies as exemplified by the abundance of their condensed liquid and solid phases Dalladay-Simpson; Dzyabura. The resultant phase diagram is complex, and the catalogue of observed phases - especially of the solid ones - steadily increases Dalladay-Simpson; Howie. The lack of clarity concerning their crystal structure in many cases is intimately connected with an incomplete insight into their electronic properties. However, it has been unclear until very recently Dias whether the solid-hydrogen atomic and metallic phase may indeed exist. Nonetheless, the detailed nature of this transition from an insulating molecular phase to the (quasi-)metallic atomic state, is still under debate McMinis; Drummond, starting from the historic paper by Wigner and Huntington Wigner. Once confirmed EremetsDrozdov, the recent work Dias would represent a decisive step in achieving our understanding of the metallization of molecular hydrogen both experimentally and theoretically. The fundamental question is whether this transition is of the Mott-Hubbard type, i.e., driven by the interelectronic correlations Mott; Gebhard or is it in class of general dielectric–metal transition driven simply by the formation of overlapping bands under strong pressure Landau; Landau2. The principal purpose of the present paper is to provide an affirmative answer to the former possibility, albeit limited to a two-dimensional situation.
Our discussion of the problem is based on an original method of approach, so it is proper to sketch first the context of the current theoretical methods applied. Many, if not most of the attempts performed up to now are based on the* Density Functional Theory* (DFT) approach. However, as it was reported by Azadi et al.Azadi, the results coming from the DFT are often ambiguous and depend strongly on a selection of the form of the *correlation-exchange * potential. Furthermore, obtaining a proper asymptotic behavior (i.e., the value of the dissociation energy) for the molecule in the large-intermolecular-separation limit is also questionable, or at least not straightforward within that approach. Whereas a proper description of the dissociation is crucial for the proper description of the metallization, as well as for the molecular crystal stabilization by taking into account the long-range London dispersion forces, a proper account of the electron-electron correlations is regarded by us as equally important. Also, the DFT-based methods such as LDA+U, LDA+DMFT suffer from the so-called double-counting problem, making their usability questionable for these systems, where the interelectronic correlations play the crucial role, particularly for low-dimensional systems. In this work we apply a specific, in principle rigorous method called the EDABI (Exact Diagonalization Ab-Initio method) which allows to surpass the last difficulty Kadzielawa; Biborski; Kadzielawa2; Spalek. However, the scope of this work is more general. Namely, we treat carefully the interelectronic interactions in the second quantization scheme and concurrently readjust variationally the single-particle wave functions, contained in the microscopic parameters, when constructing the resultant system correlated state. This method of approach thus inverts the order of executing the whole program of determining the electronic properties by diagonalizing the Hamiltonian including interactions in the second-quantization language and determining concomitantly the single-particle wave functions. Also, the present work is an essential extension of our recent communication Kadzielawa on quasi-one-dimensional hydrogen ladder to the two-dimensional () situation. Namely, we provide details of both the general methodological aspects of our approach and the concrete results for the stack of molecules (depicted schematically in Fig. 1). We map the whole problem onto the extended Hubbard model in which we additionally include the long-range (intermolecular) nature of interaction between electrons. From this point of view, we investigate the physical properties in an exact manner within the decomposition of the whole system into periodic units, each containing molecules. In particular, we focus on the Mott-Hubbard physics of the system by generalizing it to the situation when an insulating and diamagnetic molecular solid transforms into a paramagnetic atomic and metallic bilayer of atoms.
The structure of the paper is as follows. In the following Section we provide description of the applied methodology and detail the model. Next, we discuss the phase transition induced by an external side force (effective pressure) and relate it to that of the Mott–Hubbard transition for correlated systems. Finally, we discuss a possible extension of the method to the three – dimensional () systems which represent a final, not yet achieved goal within our method.
II Method: Exact Diagonalization - Ab Initio approach (EDABI)
Our methodology of approach is based on the variational approach which is an extension of the elaborated earlier in our group Exact Diagonalization Ab Initio (EDABI) scheme in the following manner Kadzielawa; Biborski; Kadzielawa2; Spalek. EDABI combines both the first- and the second-quantization schemes. What is fundamentally important, in this work we go both beyond the parametrized-model methodology Hubbard; Fazekas; Fulde and put the emphasis first on the interelectronic correlations and simultaneously renormalize the single-particle wavefunctions when constructing the resultant correlated state. To achieve this goal we start with the general electronic Hamiltonian in a second-quantization form representing an interacting system of fermions Fetter, i.e.,
[TABLE]
Hamiltonians in the first (canonical) quantization are for single () and pair of particles () respectively. and are the field operator and its adjoint, respectively. By introducing fermionic creation and anihilation operators ( and ), conforming the usual anticommutation relations
[TABLE]
where denotes spin variable, the field operators can be represented by an expansion in the creation(anihilation) operators, weighted with the amplitudes which represent single-particle wave functions forming a complete and orthogonal basis in the Hilbert space, i.e.,
[TABLE]
Hamiltonian (1) consists of one–electron part associated with the Hamiltonian for a single particle
[TABLE]
where refers to the coordination of atomic centre and is the number of sites, and of the electron-electron interaction part
[TABLE]
In both equations we used atomic units (a.u). Combining equations (1) and (3) leads to the Hamiltonian expressed in the language of creation and annihilation operators in the usual form
[TABLE]
where and are one- and two-electron interaction parameters defined as
[TABLE]
In the computationally tractable scheme expansion (3) is truncated, i.e., the sum in (3) is assumed as finite. Additionally, the functions in the expansion have their own, or may be supplied with, internal parameters , in addition to the quantum numbers characterized by the set . These parameters might be used in the variational procedure to optimize the finite-size basis composing an approximate form of , in the correlated state, i.e.,
[TABLE]
where is a finite number. In that situation,the integrals defined in (7) depend also on and, in effect, we obtain a trial Hamiltonian , for which we solve eigenequation (in our case by means of the Lanczos diagonalization method) for the many-electron problem, i.e.,
[TABLE]
where is a trial eigenvalue related to the trial many-body state. The variational procedure relies on finding the minimum of with respect to . Accordingly, the procedure is limited to relatively small systems, containing typically over a dozen electrons and corresponding to them single–particle states, providing an exact solution, at least in principle. As we have shown previously Biborski, the calculation of integrals (7) can be expensive in terms of the computational time. Below we provide the procedure of evaluating them. Note that the diagonal hopping element , i.e., the single-particle atomic energy is here also important as we discuss the system evolution with pressure which alters also the atomic energy.
III Starting system: Two-dimensional stack of molecules
We consider hydrogen molecules stacked vertically on a square (x-y) lattice (cf. Fig. 1). This molecular crystal is parametrized by the bond-length and the inter-molecular distance (lattice parameter) .
It must be stressed that even though we consider a finite system, we emulate the translational invariance by imposing the periodic boundary conditions (PBC). The supercell contains four molecules. Let us assign each molecule in the lattice by integers etc. Additionaly, we introduce the indices and to distinguish the two atoms within the molecule. Since it is assumed that single–particle states form orthogonal and normalized basis, we have
[TABLE]
where .
In this manner, each atom is labelled with the pair of the indices which also results in the labelling of the microscopic parameters, i.e., and . Effectively, we consider a degenerate two-orbital system.
Functions are approximated by means of the tight–binding approach, i.e., as a linear combination of Slater orbitals which are defined as
[TABLE]
where becomes single variational parameter to be adjusted in the correlated and stands for the atomic position, i.e.,
[TABLE]
with the summation related to extended up to the coordination zone, (c.f. Fig.2).The mixing coefficients for a given set are to fulfill condition (10) within terms of the previously elaborated procedure Kadzielawa; Biborski. Both one- and two-electron integrals (Eqs. (7a) and (7b), respectively) are also taken into account up to th coordination zone, i.e., extend beyond the supercell and in this sense we include long-range interactions. Note that subscript indices in the hopping and the interaction terms in (7) are related to the positions of the atomic centers, i.e., each pair refers to the distance. We choose the indexing in such a manner that the coordination zone number for fulfills relation . In accordance with our previous investigations Kadzielawa, we consider only the two-electron terms with the following coupling constants
[TABLE]
with when . In effect, taking into account the classical electrostatic interactions between the protons, as well as the interactions within single molecule, the total Hamiltonian describing the system is taken in the form
[TABLE]
where and . The primed summations excludes the case of concurrent and . Also, we have neglected here direct exchange-interaction terms and the additional many-site terms, as they are regarded as not essential to the physics of the problem, when considering the threshold of metallicity approached from the molecular side.
IV Computational method and physical results:
From 2D molecular crystal to quasiatomic metallic bilayer
IV.1 The enthalpy and the pressure definition in two dimensions
The system is studied here under action of an external side force (effective pressure). However, in such a two-dimensional situation the pressure has to be redefined. Namely, an external homogeneous force is exerted on the crystal in the planar (x-y) directions. Therefore, this situation is a analog of the action of hydrostatic pressure onto a three-dimensional system. The elementary volume of crystal is simply
[TABLE]
and thus, the pressure in the present case is
[TABLE]
where is the force per "unit cell" exerted homogeneously on the system in the planar directions. By taking this definition of pressure we have the usual definition of work part of the internal energy (or the enthalpy) as . Note that an infinite nature of the system is considered here preserved by means of applying PBC. Finally, a proper function of state, which in this case is enthalpy per molecule can be defined as
[TABLE]
where is the ground state energy for given structural parameters and (c.f Fig. 1). We scan the space of the parameters to obtain the energy landscape of . Note that the meaning of arises from the notion that the enthalpy should be defined as an extensive function of the system volume , where is the number of molecules in the system. Also, as an outcome of our approach, we obtain evolution of the system as a function of the applied force as the only independent variable, i.e., . In this manner, the theory is fully microscopic, as all the microscopic parameters of the Hamiltonian (14), as well as and , are determined explicitly, within our EDABI procedure.
IV.2 Computational details
The whole procedure is composed of the three stages: (i) selection and orthogonalization of the starting trial basis \big{\{}{w_{i}^{\mu}(\mathbf{r})}\big{\}}, (ii) calculation of integrals and , and (iii) diagonalization of Hamiltonian matrix and concomitant minimization of the ground state energy with respect to .
The orthogonal single particle basis is obtained in (i) in terms of the numerical solution of the bi-linear set of equations (10) with the desired accuracy ( in our case is assumed as sufficient). Step (ii) is also performed numerically by means of the previously elaborated method Biborski. Each of the Slater orbitals, which are the building blocks of functions (see Eq. 12), are approximated by three Gaussian functions what simplifies the calculation of the two-electron integrals composing Kadzielawa; Biborski. Note also that according to the spatial cutoff assumed for the repulsive Coulomb interactions, there are (intersite plus one intrasite, respectively) integrals to be computed, carried out each time when the variational parameter is updated during the minimization procedure. This stage is the most time consuming in the whole procedure. The step (iii), i.e., the Hamiltonian matrix diagonalization, is performed for the moderately sized matrix (), and results from the assumed model, i.e., that with the half filling for the -site system. The periodic booundary conditions (PBC) are imposed in the standard manner by means of inclusion of up-to-cutoff terms in the Hamiltonian matrix (cf. Fig. 2) which is diagonalized subsequently with the help the Lanczos algorithm. The diagonalization of (14) results thus in obtaining the trial value of the trial ground state energy . The latter is minimized with respect to by means of numerical procedure devoted for a single variable function numerical scheme (e.g., Brent, as in this case or golden section search), implemented within the Gnu Scientific Library (GSL) used by us in this context. The typical numerical accuracy of the energy evaluation is Ry. As the phase transition to the quasiatomic phase is of the first-order nature, such accuracy is sufficient as we can trace the evolution of the involved enthalpies in a systematic manner, as a function of applied pressure.
IV.3 Discontinuous transition and its overall characteristics
We start our discussion with remark that the solid hydrogen dissociation from molecular to the quasiatomic state, and associated with it metallization, represents one of the the fundamental transitions in Nature, as it involves one of the simplest condensed systems in which the electronic correlations play a decisive role, as we discuss next. In Fig. 3 we present exemplary results for the ground-state energy versus the bond length for the four selected values of of the lattice parameter . With the decreasing , the molecular bond length evolves from the value at ambient pressure to that close . Such a changeover speaks directly about the transition from molecular to quasiatomic configuration. The detailed character of the transformation is shown in Fig. 4, where we have displayed the enthalpies of two molecular () phases and the atomic one () as a function of applied pressure. Two discontinuous (first-order) phase transitions are seen at the critical pressures and , respectively, where is the Bohr radius. Note that at the equilibrium values of the binding energy and the bond length are and , respectively. These values can be compared with those for molecule: and Kolos. So the solid molecular bilayer is stable against the dissociation into individual molecules and the bond length in the former case is larger by . This result provides a crucial test of our method reliability when applied to the multimolecular systems. Obviously, the values of at prove only that the solid molecular phase is stable for from the electronic point of view, as we have not included as yet the zero-point motion. Those will be estimated later. The application of pressure will help additionally to stabilize it.
In Fig. 5 we plot the equilibrium lattice parameter (in units of ) versus pressure and observe a discontinuous lattice contractions for both the transitions by about and at the pressures and respectively. In an analogous manner, the bond length vs pressure jumps from the equilibrium value to at the critical pressure , as shown in Fig. 6. Hence the transitions are strongly discontinuous between the each of the two pair of three stable phases. The phase diagram for the scanned space of is composed of three phases. Those referring to and we recognize as both being of a molecular kind and label them them as phases I and II, respectively, while the phase referring to is the quasiatomic one. This distinction may seem at this stage as somewhat arbitrary and is legitimate only by making observation that the ratio for stable phase referring to and for . However, more convincing argument which originates from the diversity of electronic properties for both of the two groups of phases, is provided in the next subsection. As a supplementary information we have plotted in Fig. 7 the inverse Bohr radius vs for the Slater functions composing the Wannier functions. The jumps take place by at and by at , so the wave-functions site is strongly altered at both the transitions. Note that value in the phase is close to that for the hydrogen atom (within ) even though the actual value in the quasiatomic solid phase is only about of the single-atom value. This last results is certainly counterintuitive. Interelectronic correlations, induced by the interatomic repulsive interactions, reduce the effective Bohr radius by over in the molecular phase II.
IV.4 Principal electronic characteristics of the Mott-Hubbard transition
For the sake of completeness, we list in Table 1 principal parameters of the three states calculated at the critical pressures. Particularly interesting are and , the intra- and inter-molecular hopping integrals, since they change remarkably at the transition. The same concerns the values of the Hubbard gap (with the bare bandwidth calculated in Appendix A) and the ratio (c.f. Figs. 8 and 9, respectively). The last characteristic is particularly important since at the transition at it jumps from (), in the molecular state to the value () and represents a typical trend for the Mott-Hubbard transition, albeit this time from an originally * diamagnetic molecular insulator to a paramagnetic metal*. The negative value of the Hubbard gap means that the two lowest bands overlap appreciably and therefore the system can be regarded as metallic.Also, there is a principal difference between the present approach and the the canonical treatments Gebhard; Fazekas; Fulde of the Hubbard model, as here the value of the bandwidth changes at the transition, and in effect, the ratio is, not as one would have in all the parametrized-model considerations Hubbard2; Brinkman; Spalek2, changing in a continuous manner. Also, a relatively large value of the intersite Coulomb interactions may mean that either the spin (SDW)- or the charge (CDW)-density-wave states become a stable phase on the quasiatomic side, at least in the low-temperature range. This topic should be analyzed separately, as it is more complicated than the present analysis. Such an analysis would allow for differentiating in detail between the present transition from the diamagnetic insulator and the canonical Mott-Hubbard transition which takes place from an antiferromagnetic (Mott) insulator to either SDW or a paramagnetic correlated metal. Also, as said above, the Mott-Hubbard transition is analyzed customarily as a function of ratio changing continuously Hubbard2; Brinkman; Spalek2; Vollhardt. As our results show explicitly this is not the case, when the renormalization (readjustment) of the orbitals is taken into account in the correlated state. In this respect, our approach is fully microscopic (parameter free).
The transition can be elaborated further by calculating directly the intramolecular () and the intermolecular hopping correlation functions, both displayed in Fig. 10. Note that the value of correlation function reaches the value in the quasiatomic phase which we identify with the system metallicity. This is because this value reaches an amazing value for electrons per atom, characteristic of the uncorrelated lattice fermionic gas Spalek3. On the contrary, the value of in the molecular phases is close to zero, whereas then, both characteristic of a molecular insulator. It is amazing that so spectacular switching from an almost ideal insulator to an almost ideal fermionic gas takes place in this situation. The situation described here is in accord with an old argument of Mott Mott2 that switching to a metallic state can take place only in a discontinuous manner as a creation of a small number of carriers in a nominally insulating state would largely increase the system energy due to the lack of screening of the long-range repulsive Coulomb interaction between them. Here, this argument is fully qualified and includes also the Hubbard argument Hubbard in the same manner. In effect, the solid hydrogen may be indeed regarded as the model example of the transition from a correlated, albeit diamagnetic insulator to a moderately/weakly correlated paramagnetic metal, if we only account properly for its original molecular structure in a solid at ambient pressure, and subsequently the renormalization of both the molecular and the atomic (Slater) orbitals by the interelectronic correlations. The values of the lattice and microscopic parameters at the transitions are listed in Table 1.
IV.5 Electron density evolution and renormalized single-particle band characteristics in the correlated state
To complete our picture of the metallization we have also determined the electron densities in the many-particle states; those are displayed in Figs. 11 and 12, in both the molecular and the quasiatomic states. The nature of the states does not alter qualitatively in Fig. 11; they represent indeed the two molecular states, differing only by the bond length, etc. On the contrary, the nature of the molecular – quasiatomic transition is very clear, since the density shown in Fig. 12b splits with respect to (a) into two disjoint regions representing well separated states of atoms. The latter states are called quasiatomic because their size (cf. Fig. 7) differs remarkably with respect to that of an isolated atom.
The above evolution of electron density for molecules/atoms placed in the milieu of all other particles is supplemented with the selected relevant parameters displayed in Table 2, where we list the values at the consecutive transitions (marked withe subscript in each case): , , , as well as provide the critical values of the Hubbard ration and of the Mott criterion "", here adopted to the two dimensional () case, for which the effective Bohr radius is and the particle density . Those three quantities are listed in the last two columns. It is amazing that those two sets of values, introduced via a rough estimates are not far off from the standard estimates Mott at the transition to the quasiatomic state.
In Fig. 13a-b we have determined the two lowest bare-bands dispersion relations calculated with the renormalized hoppings parameters and at the transition from the molecular phase I to II, as well as in Fig. 13c-d at the transition from phase II to the quasiatomic phase. As the interactions are not included in those calculations, we do not have the Hubbard-subband structure at the transition from I to II. Nevertheless, since then , the structure represents that of an insulators, whereas the quasiatomic phase is metallic, as there is an appreciable band overlap is that state and the correlations are moderate to weak (). The phases I and II are both insulating; they differ only by different values of the microscopic parameters. It is tempting to suggest that while the phase I is diamagnetic, the phase II may be of insulating and (antiferro)magnetic. However, this point requires a separate analysis.
To provide an illustrative evidence for the existence of two distinct molecular phases, the corresponding to them enthalpy minima at those two transitions have been visualized in Fig. 14a-b. We see that even in there are two states and this circumstance may be regarded as a precursory effect for a number of such phases appearing in experiment on systems Dalladay-Simpson; Drummond.
To illustrate the changes in the single-particle functions at the transitions, we have drawn the Wannier functions at the I→II- (cf. Fig. 15) and II→quasiatomic-state (cf. Fig. 16) transitions along in-plane ( - (a)) and molecular ( - (b)) directions. The two equilibrium lattice and bond parameters have been supplied in each of the Figures. Their evolution reflects perfectly the trend of the Slater-orbital size () jumps shown in Fig. 7. It is amazing that they look more atomic-like in the last, metallic phase. However, the situation is not so simple, since at the same time the lattice parameter decreases appreciably in a discontinuous manner at the same time and therefore the change of Hamiltonian parameters is also influenced by that. Nonetheless, the bond-length changes are most important (cf. Fig. 14.
Concluding this Section, the results presented in Figs. 9, 8, 10, 11, 12, 13, 14, 15 and 16 provide an unequivocally evidence for the molecular to quasiatomic phase transition at the critical pressure . Obviously, a further evidence of metallicity in the latter phase would require a direct calculations of the electric conductivity. Namely, it would require an extension of the present approach to nonzero temperature, as here the conductivituy at would take the values in the molecular phases and in the metallic ground state. However, the gap closure at the II → quasiatomic discontinuous phase transition (cf. Fig. 13) provides a clear sign of metallicity in the latter phase.
V Outlook
V.1 Brief summary and zero-point motion of atoms
Let us summarize first our effort here. We have discussed the metallization of stack of molecular hydrogen within the EDABI method. The method relies on an exact diagonalization of the Hamiltonian describing the dynamic processes within supercell containing molecules; this cluster is subsequently repeated periodically in the both planar directions, with additional inclusion of the hopping and interelectronic interactions extending beyond the supercell (cf. Fig. 2). In this respect, our approach represents a version of coupled-cluster approach Maska. Furthermore, at each step of the iterative diagonalization procedure by Lanczos method we readjust the single-particle (Wannier) wave function until a fully microscopic ground-state energy configuration is reached. Therefore, the input parameters are solely the atomic structure (cf. Fig. 1) and the finite single-particle basis, limited here to states. As a results we obtain the principal characteristics such as the lattice constant, the effective bond length, renormalized band structure and single-particle wavefunctions, and the ground-state enthalpy, all as a function of applied force. But first and foremost, we obtain the sequence of discontinuous phase transitions and in particular, the insulator – metal transition from the insulator to metal. The atomization process is illustrated directly in Figs. 12, where, the many-particle electron-density profiles have been drawn. All this provides the evidence that the hydrogen metallization represents a transition of the Mott-Hubbard type, though the starting material at ambient pressure is a diamagnetic (not antiferromagnetic) insulator. Hence, our approach represnts an essential extension of the concept of the canonical Mott-Hubbard transition.
Our analysis would be complete if we have supplemented the present work with the study of stability of the assumed protonic lattice in the metallic state. In other words, a separate question can be asked if the metallization is not associated with the transition to a liquid proton-electron plasma state Dzyabura; Weir; Tamblyn; Morales; Silvera, though the corresponding transition in the liquid is also observed Nellis2015. The latest experimental results Dias support the view taken here that the lattice survives the strong first-order transitions (see however Ref. Eremets). The stability of the lattice can be justified by two features of our results. First, we have shown that at the transition the electron orbit shrinks remarkably (cf. Figs. 7 and 16), screening the charges on a short-distance scale and thus diminishing the repulsive energy. Second, in Appendix B we have estimated the amplitude and the energy contribution of the zero-point motion in the harmonic approximation Spalek; Kadzielawa3 and both in the inter- and intra-molecular driections. The inclusion of the zero-point motion can change only slightly the transition points without changing the overall features of our results. Note that the ZPM energy does not exceed of the total enthalpy value (cf. Fig. 4), but up to about of the ground-state-energy value.
V.2 Relation to other works
The principal novelty of our approach relies on: (i) implementing a combined first- and the second-quantization scheme which allow for a full ab-initio analysis of the correlated state and without the appearance of the notorious double-counting problem; (ii) determining the renormalized Wannier orbitals which supplement the whole picture qualitatively with respect to that obtained within the parametrized models; and (iii) applying the concepts of the Mott-Hubbard transition to the canonical solid hydrogen system with orbitals. The transition is accompanied by a simultaneous two-step transition from the correlated diamagnetic molecular insulator to the two-dimensional metal as a function of applied pressure. The inclusion of long-range Coulomb interaction should also be noted. The question remains as to whether such a bilayer system can be realized experimentally by e.g., covering a substrate with a plane of such stacked molecules, with the substrates of variable lattice parameter emulating the pressure applied to the system edges. Such an experiment could provide a direct realization of a bilayer crystal in a metallic state. In this case of a bilayer deposited on the substrate one would have to account also for the dynamics of the protons and electrons in the presence of a trapping them external (surface) potential which, if sufficiently strong, would suppress their zero-point vibrations.
Other theoretical works involve among others recent diffusion quantum Monte-Carlo simulations Azadi2; Drummond and advanced DFT methods McMahon; Kudryashov. Both methods predict the metallization for pressures in the range in the three-dimensional case. Here we show that the transition in the bilayer case is of the Mott-Hubbard type. The same type of the transition has been shown to exist in one-dimensional case Kadzielawa for the molecular ladder. Therefore we expect that the same type of results can be expected in the case, but a proof of that hypothesis requires more involved approach and should employ the incorporation of the Monte-Carlo methods into our scheme. The reason why our results are to certain degree independent of the lattice dimensionality is the fact that we include long-range Coulomb interactions between the electrons which make the results look more like those of mean field theory of those correlated fermion systems, results of which are only weakly dependent on the system dimensionality.
V.3 Concluding results
So far we have analyzed the normal states only. As our bilayer metallic phase represents a moderate correlated system we can treat it as a bilayer system with correlation-driven pairing, analogously to our recent approach of the cuprates within the extended Hubbard model Spalek4; Zegrodnik; Kaczmarczyk. However, the situation is not that simple as here the electron-lattice interactions can be quite strong, as one can see already on the and examples Kadzielawa3. In effect, both the correlation and electron-lattice parts should be treated on equal footing. In that case, one can estimate their relative contributions to the superconducting critical temperature and in such a manner complement the estimates based purely on the electron-lattice contribution Kudryashov; Ashcroft; Borinaga. We should be able to see a progress along these lines in the near future.
VI Acknowledgments
The work was financially supported by the National Science Centre (NCN), through Grant No. DEC-2012/04/A/ST3/00342. The computations have been performed in part on the supercomputer TERA-ACMiN AGH and partly on the supercomputer EDABI located at the Jagiellonian University.
Appendix A Bare bandwidth W of the electrons in the correlated state
To compute ratio, the bandwidth can be obtained from diagonalization of the single–electron part of Hamiltonian (14), i.e.,
[TABLE]
In accordance with the translational invariance of the system in the x-y plane, ( 18) can be rewritten in the momentum () representation in the form
[TABLE]
where primed summation refers to , index enumerates molecules in the assumed neighborhood, i.e., . The functions and map to proper indexing of the hoppings. Note that one may select where are integers, as we did in our considerations. In effect, the single-electron Hamiltonian can be recast in the matrix form for each spin, i.e.,
[TABLE]
Diagonalization of matrix provides the bare dispersion relation . For our molecular crystal two, spin-degenerate, branches and appear. The matrix is constructed in a straightforward manner for particular , i.e., by computing numerically with up to the coordination zone. Subsequently, is diagonalized and the two eigenvalues , are obtained. For the half filling considered here, only band is occupied by electrons. Therefore is defined in a standard manner, i.e.,
[TABLE]
Both the maximal and the minimal values, and , are obtained numerically for by scanning the eigenvalues in the first Brillouin zone. Those values were used when plotting and in Figs. 9 and 8 in main text. Note that in the molecular state the lower band is nominally filled, whereas in the metallic state the bands overlap.
Appendix B Assessment of zero-point motion in harmonic approximation
We estimate the zero-point motion (ZPM) of our system by introducing a ion position uncertainty
[TABLE]
then by splitting the problem into two parts: (i) ZPM in a molecule (), (ii) ZPM of a molecule in the crystal field (). In both cases the kinetic energy of the molecule is
[TABLE]
where , , and is approximated via the Heisenberg uncertainty principle
[TABLE]
hence .
The potential energy is calculated separately for the cases (i) and (ii).
B.1 ZPM for molecule
We base our approach by our earlier work Spalek; Kadzielawa3. We define the potential
[TABLE]
where is the energy of the molecule of the molecular size , and , the minimum of energy for static molecule (so that our potential used static equilibrium as a reference point).
The energy gain from the ionic movement is given by an expression
[TABLE]
For the given molecular size we minimize expression (26) with respect to .
B.2 ZPM per molecule in the crystal
For the case of the molecule in the crystal field we assume that the electrons do not contribute to the ionic potential, hence
[TABLE]
where is the intermolecular distance, is the molecule size, is the charge of a hydrogen ion, interaction cell refers to the molecules we considered as our background (cf. Fig. 2 for the background considered in this paper) at the positions , and is the potential of static molecules
[TABLE]
The energy gain from the ionic movement is given by an expression
[TABLE]
For the given intermolecular distance and molecular size we minimize expression (29) with respect to and .
B.3 Numerical results at the transitions
In Table 3 we present both absolute and relative magnitude of ZPM, as well as all the possible modes with their corresponding energies (in Rydberg per molecule).
