Half-metallicity versus Symmetry in Pt, Ni and Co-based Half Heusler Alloys: A First-principles Calculation
Madhusmita Baral, Aparna Chakrabarti

TL;DR
This study uses first-principles calculations to explore the stability, electronic, and magnetic properties of Pt, Ni, and Co-based half Heusler alloys, revealing the relationship between symmetry and half-metallicity.
Contribution
It provides a comprehensive analysis of various crystal symmetries and predicts new non-cubic half Heusler alloys with high spin polarization.
Findings
Only 18 out of 108 studied structures are energetically stable cubic phases.
Alloys with group VIA C atoms are often stable, especially with high atomic number elements.
A potential one-to-one relationship between cubic symmetry and half-metallicity is suggested.
Abstract
Using first principles calculations based on density functional theory, we study the geometric, electronic, and magnetic properties of Pt, Ni and Co-based half Heusler alloys, namely, Pt, Ni and Co ( = Cr, Mn and Fe; = Al, Si, P, S, Ga, Ge, As, Se, In, Sn, Sb and Te). We calculate the formation energy of these alloys in various crystal symmetries, which include, the (face-centered) cubic (3m), orthorhombic (), as well as hexagonal ( and ) structures. It has been observed that out of all the 108 structures, studied here, energetically stable cubic structure is observed for only 18 materials. These alloys are primarily having either a atom or an atom with a high atomic number. We also observe that along with the alloys with atoms from group IIIA, IVA and VA -- alloys with atoms from group VIA are also…
Click any figure to enlarge with its caption.
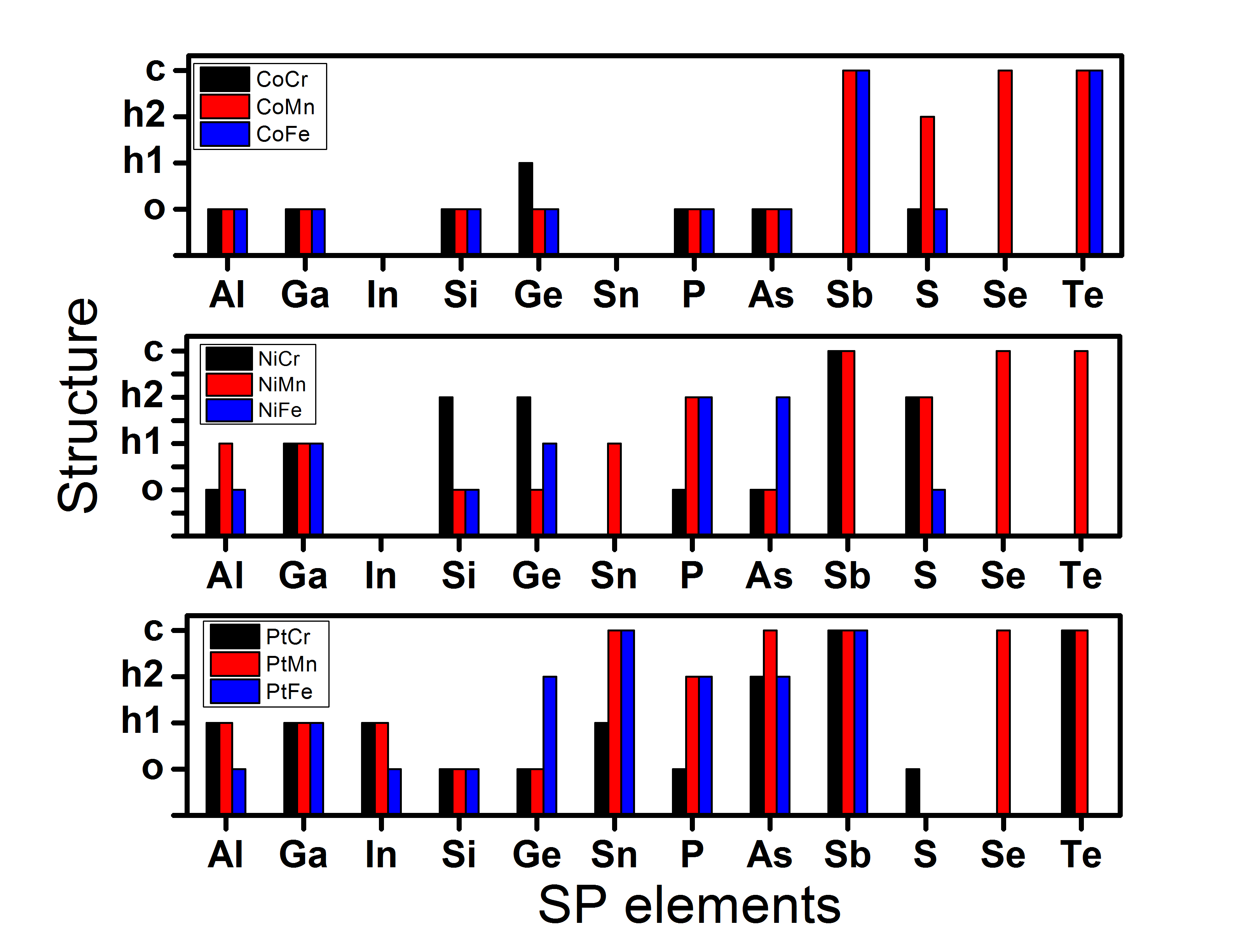 Figure 1
Figure 1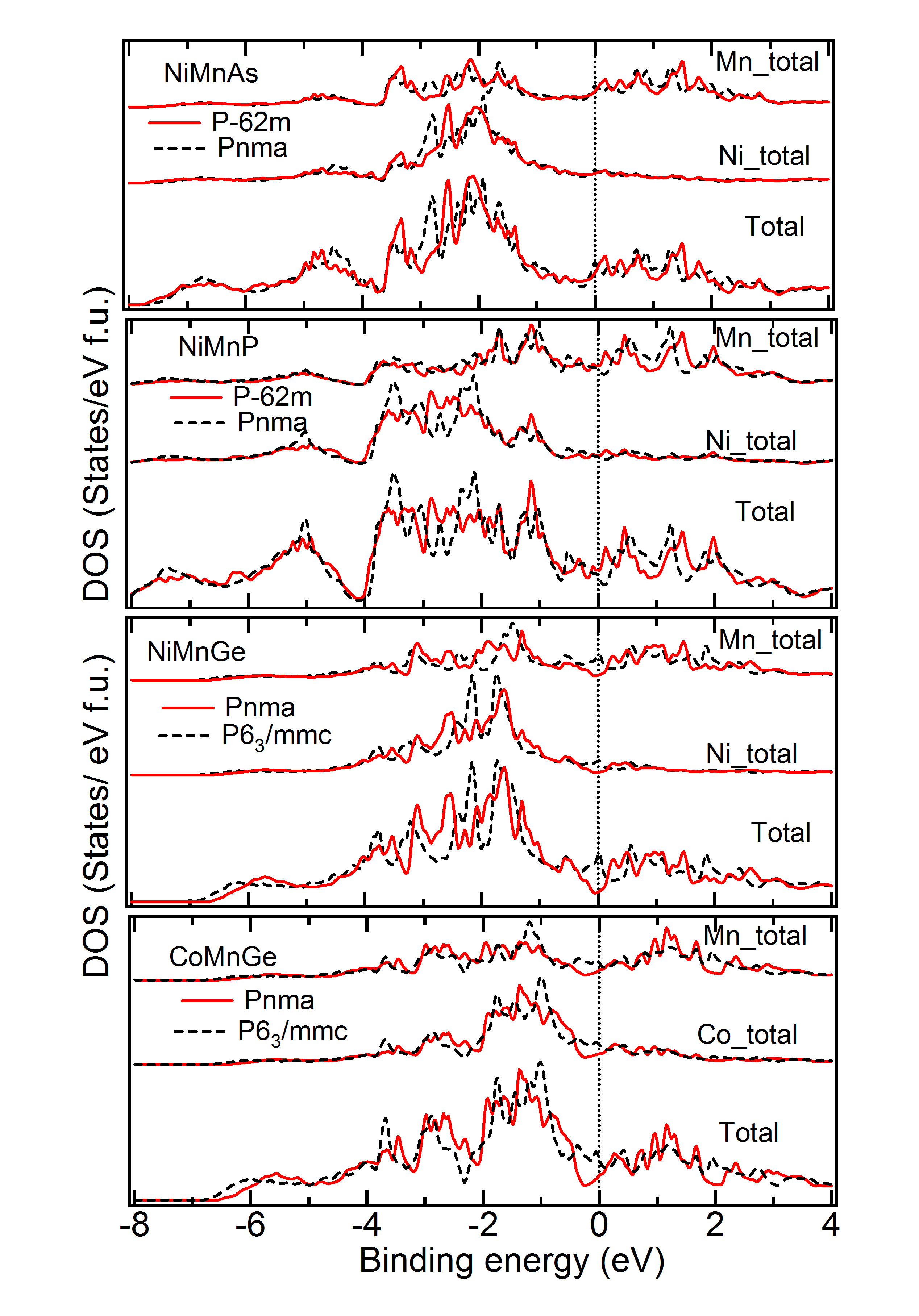 Figure 10
Figure 10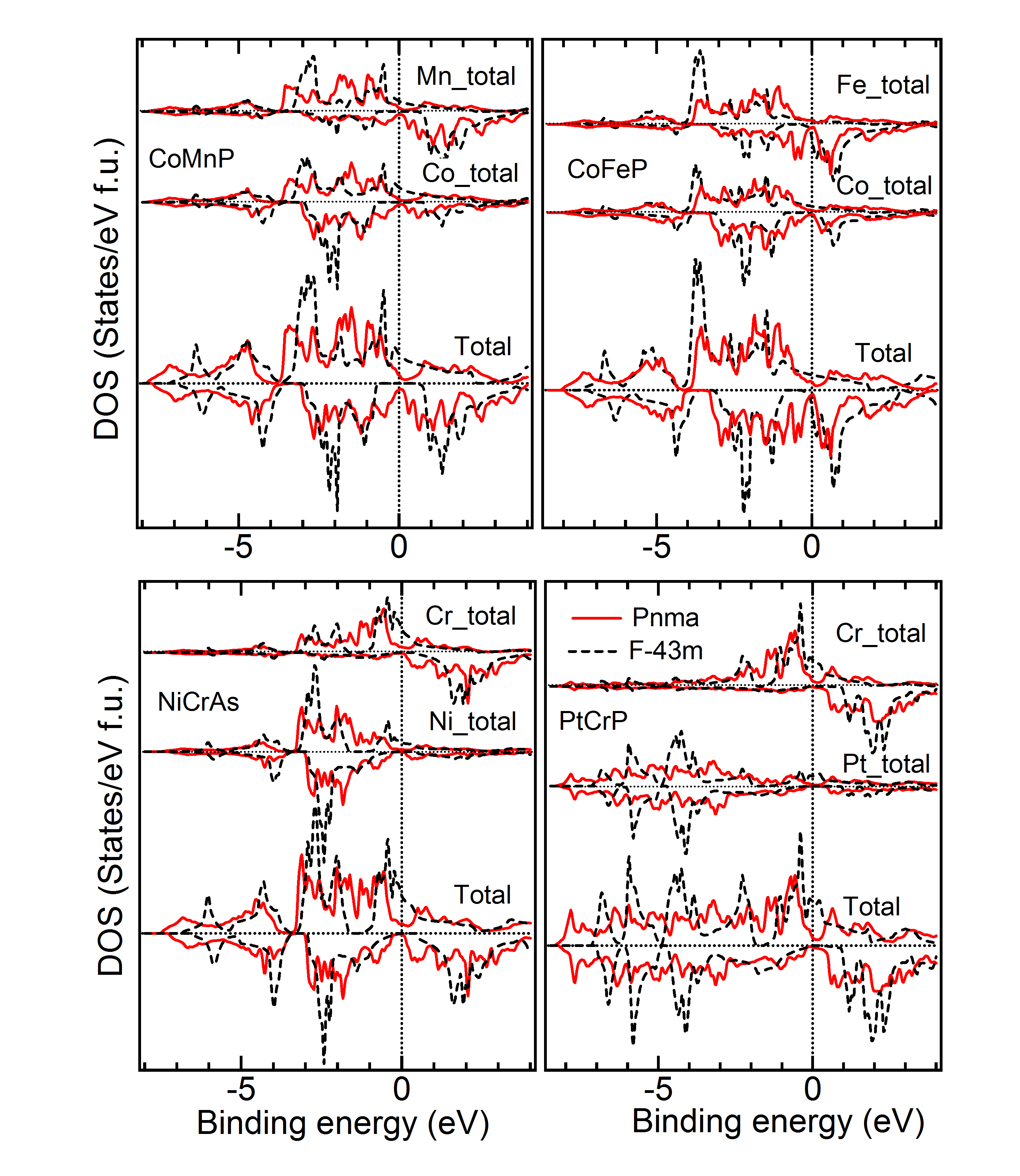 Figure 11
Figure 11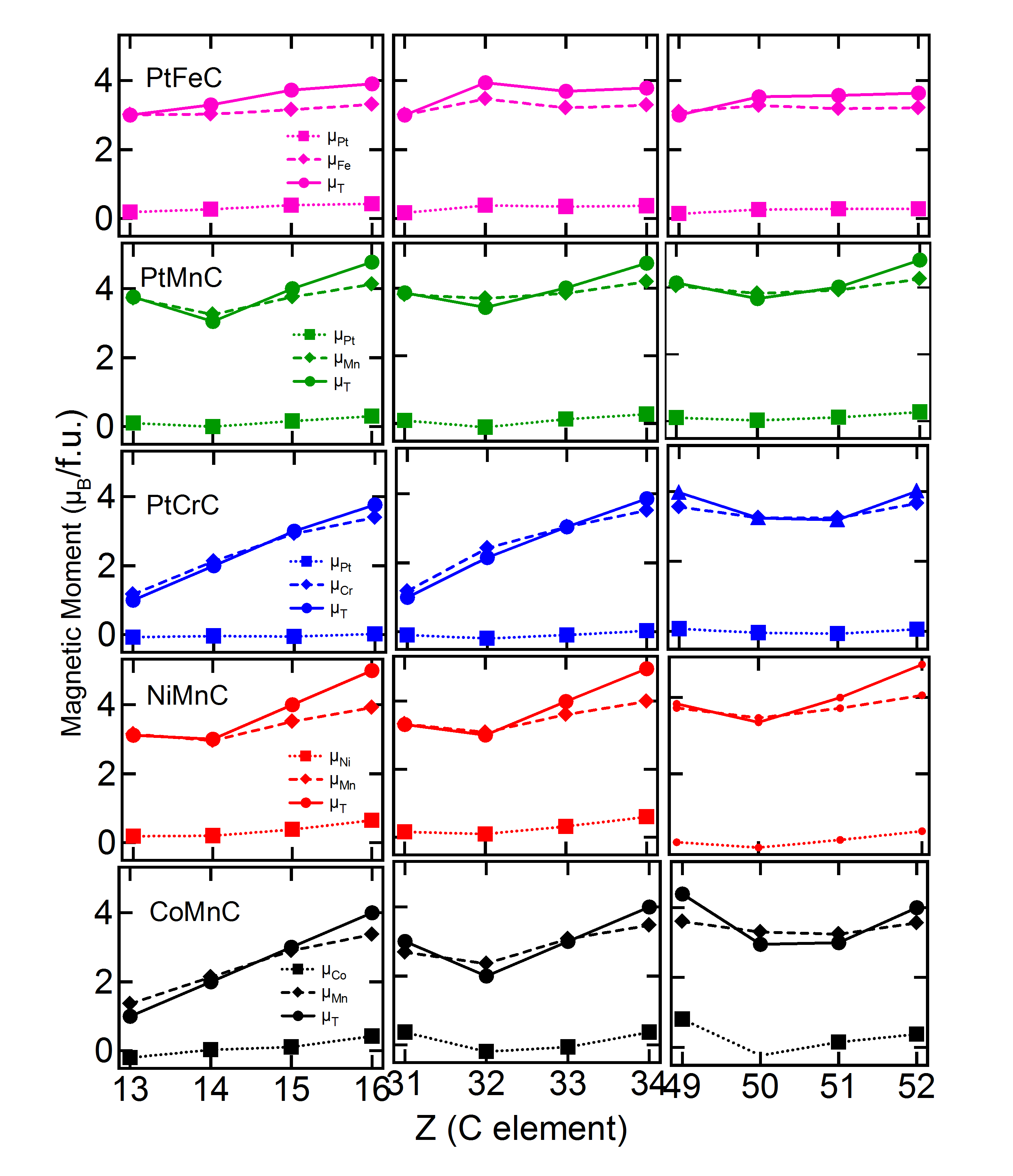 Figure 2
Figure 2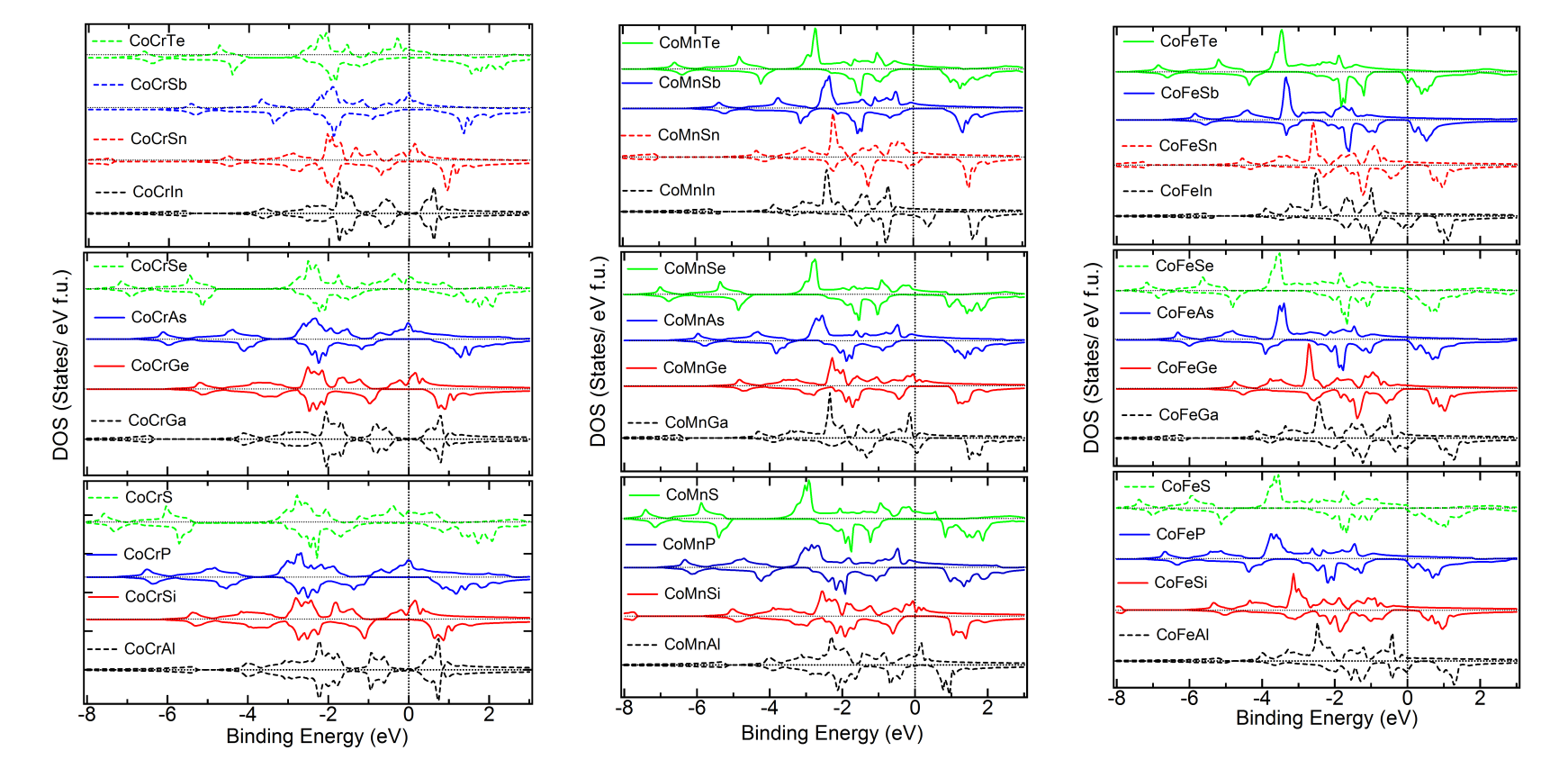 Figure 3
Figure 3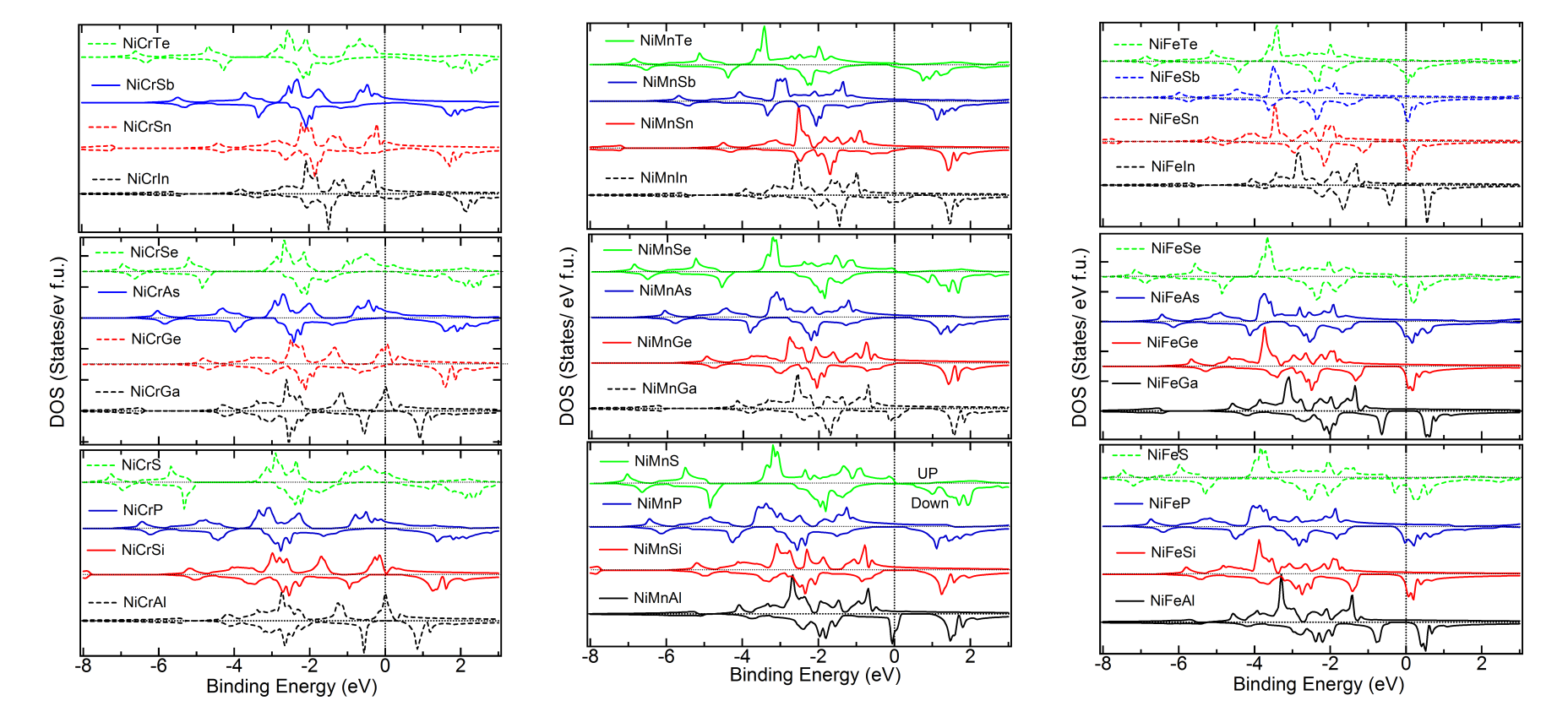 Figure 4
Figure 4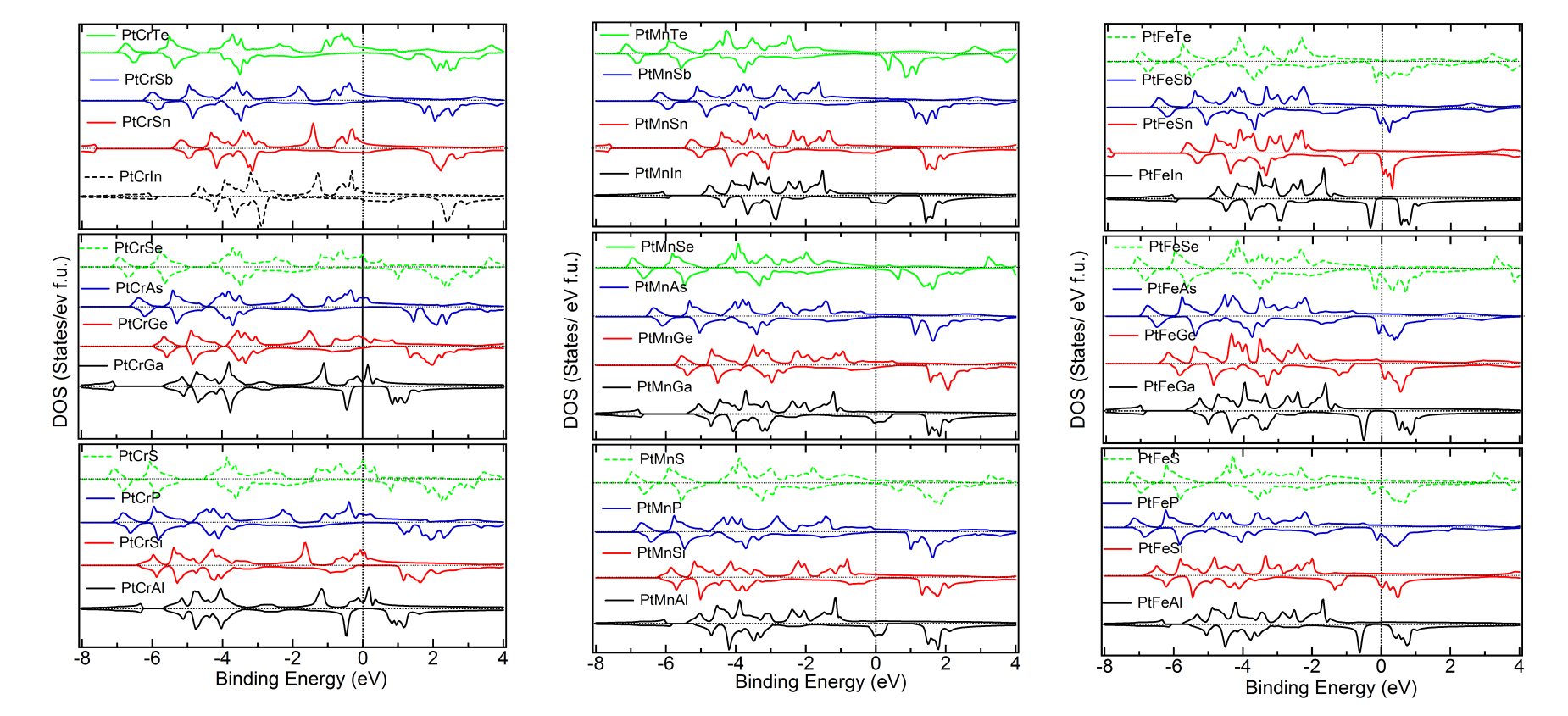 Figure 5
Figure 5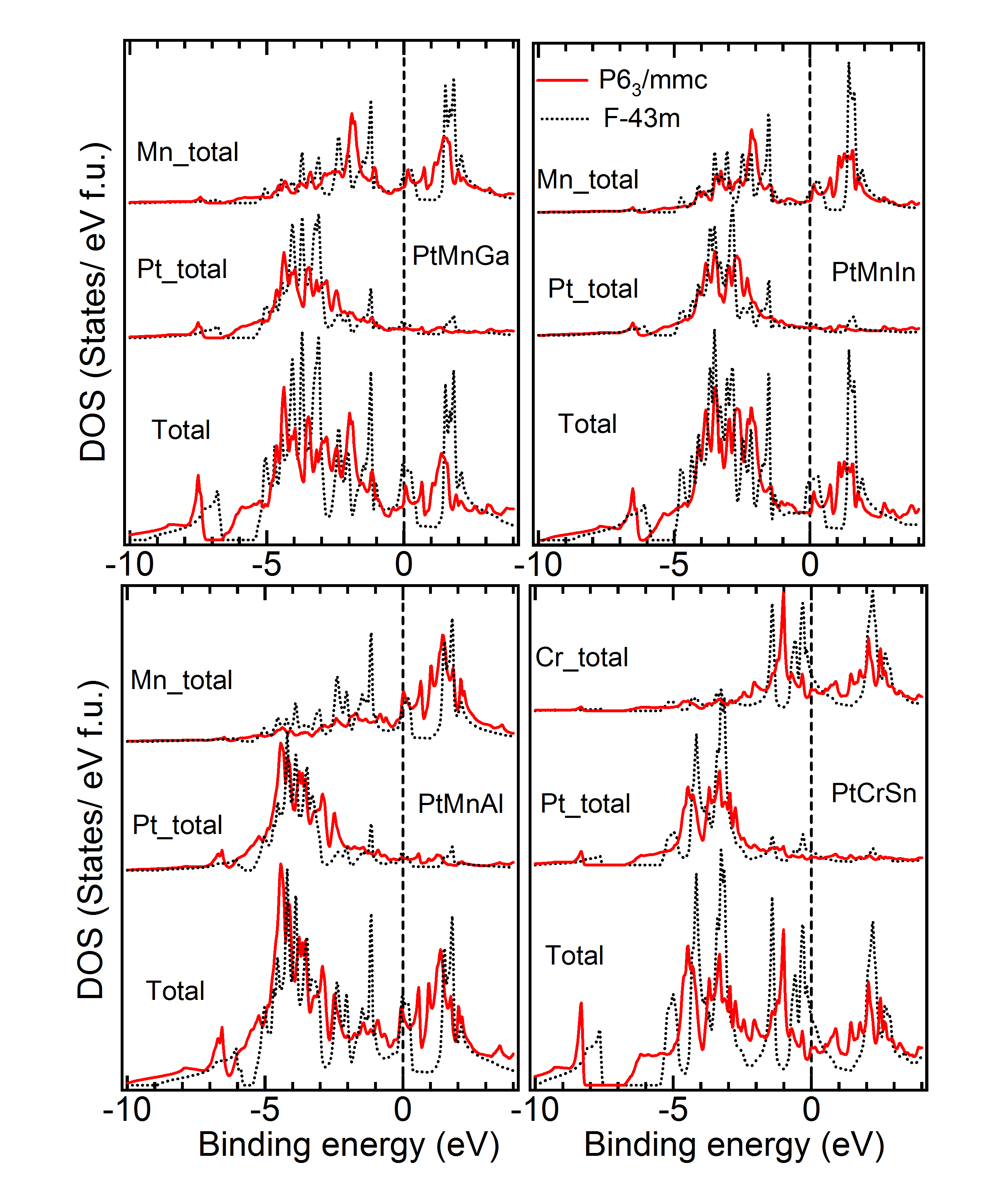 Figure 6
Figure 6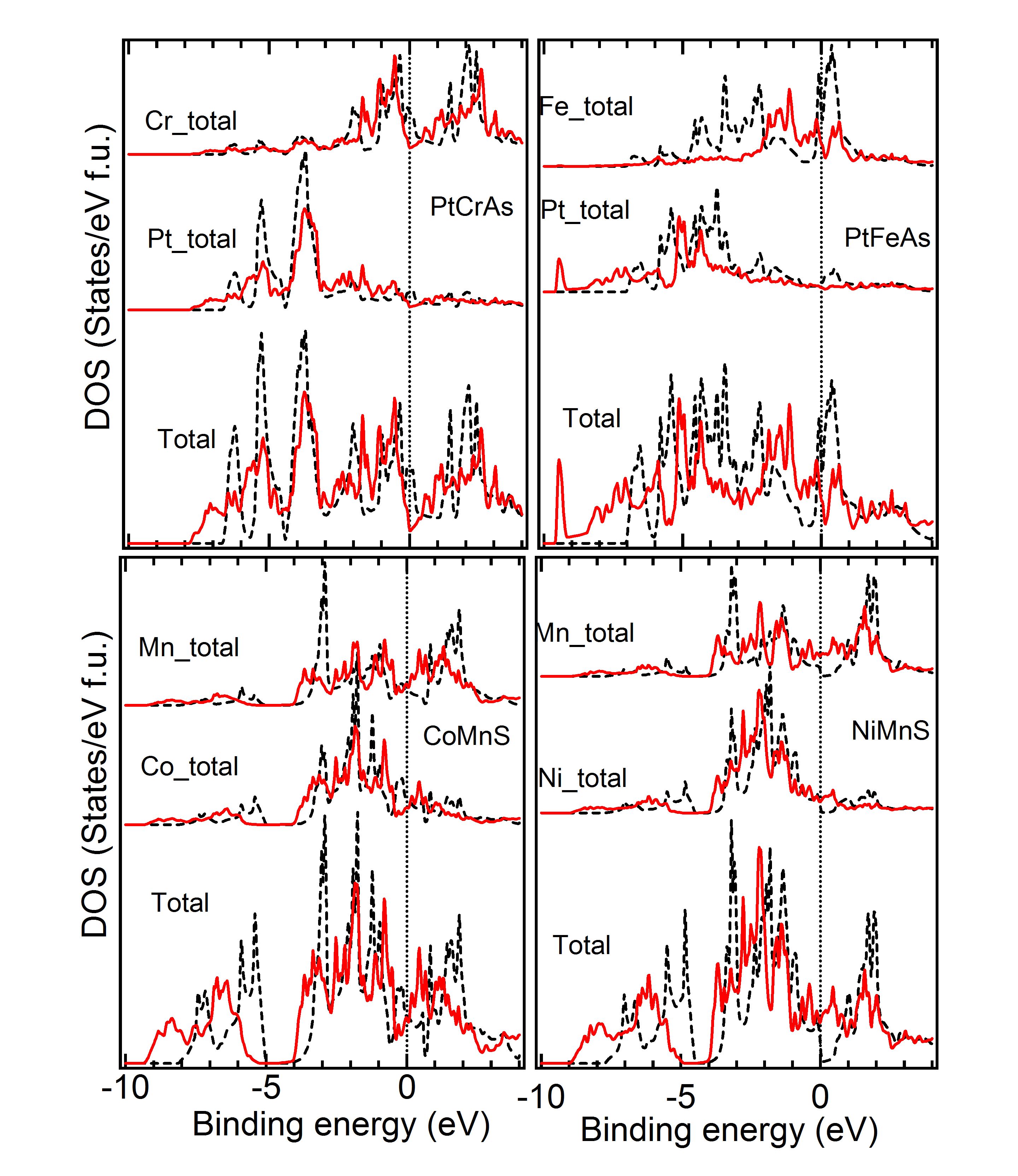 Figure 7
Figure 7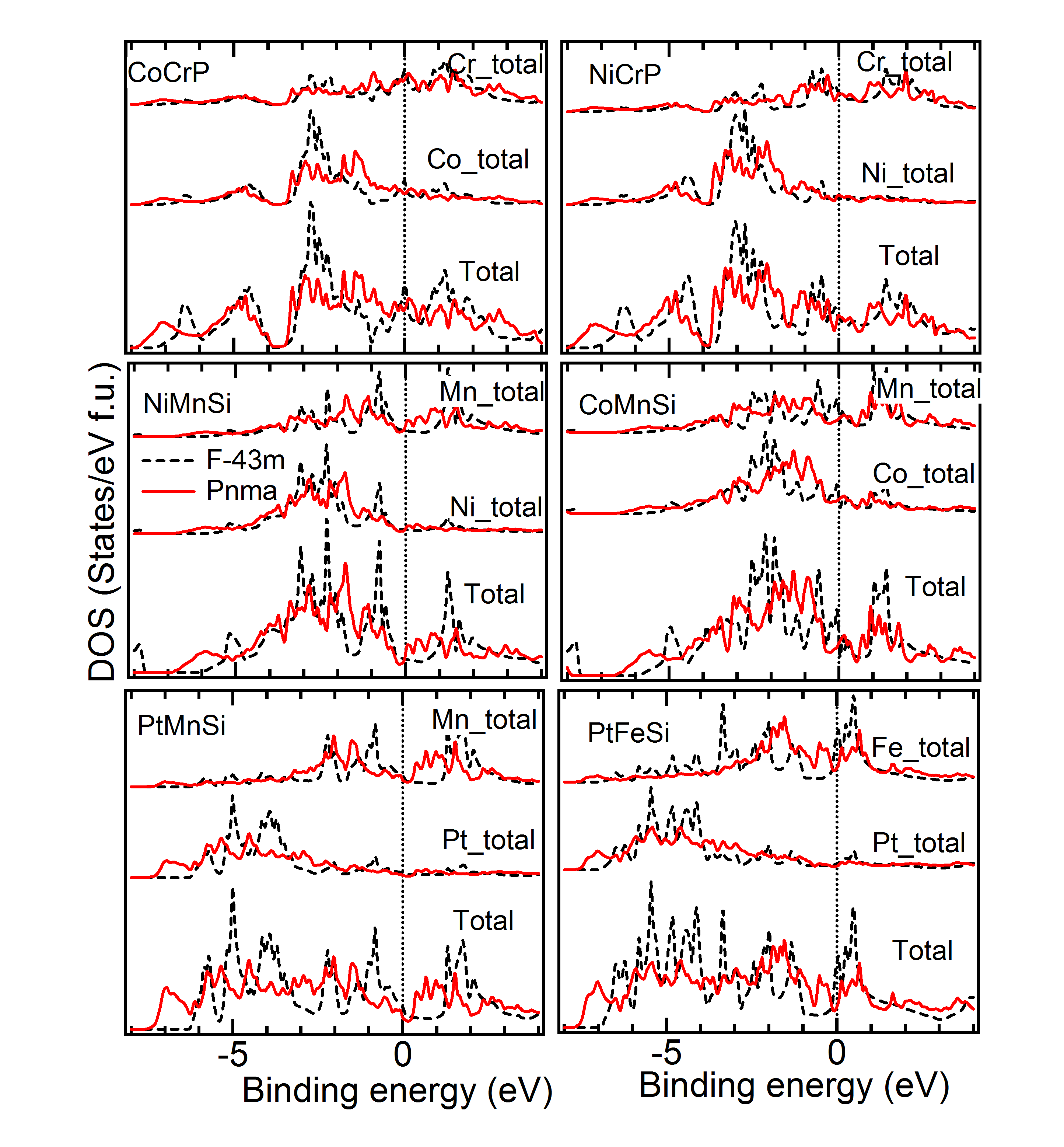 Figure 8
Figure 8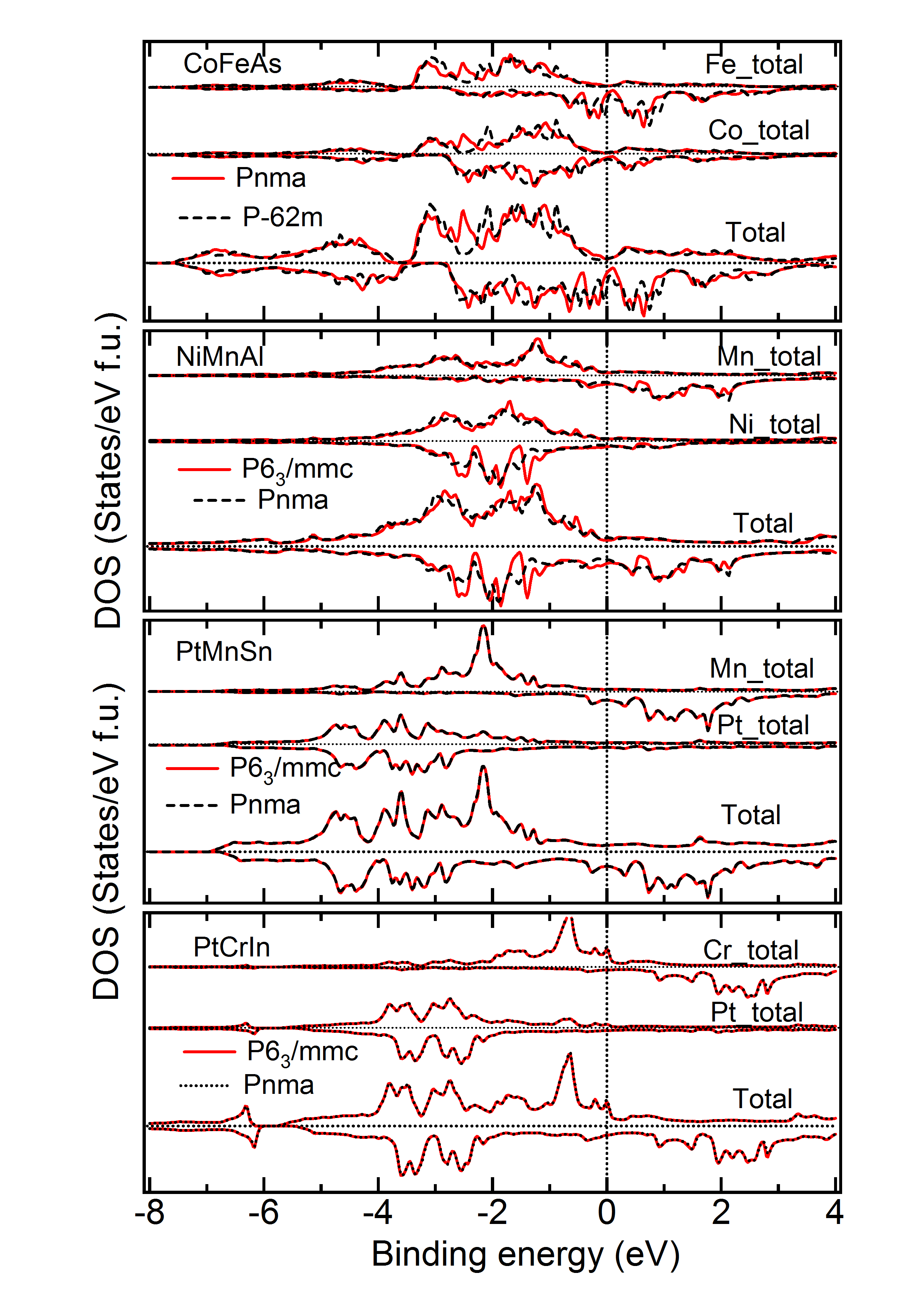 Figure 9
Figure 9Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Half-metallicity versus Symmetry in Pt, Ni and Co-based Half Heusler Alloys: A First-principles Calculation
Madhusmita Baral1,2 and Aparna Chakrabarti2,3
1 Synchrotrons Utilization Section, Raja Ramanna Centre for Advanced Technology, Indore - 452013, India
2 Homi Bhabha National Institute, Training School Complex, Anushakti Nagar, Mumbai-400094, India
3 Theory and Simulations Laboratory, HRDS, Raja Ramanna Centre for Advanced Technology, Indore - 452013, India
Abstract
Using first principles calculations based on density functional theory, we study the geometric, electronic, and magnetic properties of Pt, Ni and Co-based half Heusler alloys, namely, Pt, Ni and Co ( = Cr, Mn and Fe; = Al, Si, P, S, Ga, Ge, As, Se, In, Sn, Sb and Te). We calculate the formation energy of these alloys in various crystal symmetries, which include, the (face-centered) cubic (3m), orthorhombic (), as well as hexagonal ( and ) structures. It has been observed that out of all the 108 structures, studied here, energetically stable cubic structure is observed for only 18 materials. These alloys are primarily having either a atom or an atom with a high atomic number. We also observe that along with the alloys with atoms from group IIIA, IVA and VA – alloys with atoms from group VIA are also found to be, by and large, energetically stable. To examine the relative stabilities of different symmetries in order to search for the respective lowest energy state for each of the above-mentioned systems, as well as to find whether a material in the ground state is half-metallic or not, we analyze the formation energy, and the electronic density of states, in detail. Based on these analyses, the possibility of existence of any one-to-one relationship between the cubic symmetry and the half-metallicity in these half Heusler alloys is probed. Subsequently, we predict about the existence of a few new non-cubic half Heusler alloys with substantially low density of states at one of the spin channels and reasonably high spin polarization at the Fermi level.
pacs:
71.20.Be, 71.15.Nc, 71.15.Mb, 75.50.Cc
I Introduction
The prediction and development of new half-metallic ferromagnets are of immense interest due to their potential for technological application.generalintro The half-metallic (HM) ferromagnets (FM) are a type of FM material where spin polarization at the Fermi level (EF) is high (expected to be 100%) which may have an application in the field of spintronics. Ever since the half-metallicity has been predicted, in half Heusler alloys (HHA), namely, NiMnSb and related isoelectronic compounds, PtMnSb and PdMnSb, on the basis of band structure calculations,NiMnSbasHM the field of half-metallic half Heusler alloys (HM HHA) has attracted the interest of the researchers.PRB95JM Here we wish to point out that, further on, we refer to all the materials with high (about 65 to 70%) to 100% spin polarization, as HM-like materials.
Many HM-like materials have been found theoretically, among the half and full Heusler alloys.NiMnSbasHM ; PRB93TR ; JMMM423TR The Curie temperature (TC) of quite a few of these alloys is calculated to be higher than the room temperature which is essential for their application as an efficient and useful spin-injector material.highTCHM For this application, the interest is indeed in magnetic HHAs. It has been seen that a large amount of work on NiMnSb and substitution at its different atomic sites has been carried out, both theoretically and experimentally.NiMnSbasHM ; workonNiMnSb In order to search for new half-metals, a large number of general type HHAs have also been studied in the literature, where and are transition metal atoms and is an sp element.workonABC
In the literature, most of the HHAs studied theoretically have been shown to possess half-metallic property in the cubic C1b phase with F3m space-group.NiMnSbasHM However, only some of these have been experimentally synthesized.exptHMHHAcubic On the contrary, it has been seen in the literature that many of the half Heusler alloy samples exhibit non-cubic symmetries but there is no explicit discussion on the half-metallicity in these materials.workon-non-cubic-Co+Ni-Cr+Mn+Fe-Si ; workon-non-cubic-Co-Cr+Mn+Fe-P+As++Ni-Cr+Mn+Fe-P+As ; workon-non-cubic-Co+Ni-Cr+Mn+Fe-P++Co+Ni-Cr+Mn-As ; workon-non-cubic-CoCrGe ; workon-non-cubic-Co+Ni-MnGe ; workon-non-cubic-CoMnGe++Co+Ni-FeGe ; workon-non-cubic-Co+Ni-Mn+Fe-P ; workon-non-cubic-CoMnAs ; workon-non-cubic-CoMnSb ; workon-non-cubic-NiCrP ; workon-non-cubic-NiMnGa ; workon-non-cubic-NiMnGe ; workon-non-cubic-Pt-Cr+Fe-Sn ; workon-non-cubic-PtMnGe++PtCrSb ; JMMM38 It has been observed that NiMnSb and some other Sb, as well as Sn materials possess cubic C1b symmetry.JMMM38 On the other hand, while NiMnGe is seen to exhibit an orthorhombic space-group at room temperature,JMMM27 NiMnGa has a hexagonal symmetric structure.workon-non-cubic-NiMnGa Further, Si or Ge or P or As compounds are seen to exhibit either an orthorhombic structure (with space-group) or a hexagonal phase (with either or space-group).workon-non-cubic-Co+Ni-Cr+Mn+Fe-Si ; workon-non-cubic-Co-Cr+Mn+Fe-P+As++Ni-Cr+Mn+Fe-P+As ; workon-non-cubic-Co+Ni-Cr+Mn+Fe-P++Co+Ni-Cr+Mn-As ; workon-non-cubic-CoCrGe ; workon-non-cubic-Co+Ni-MnGe ; workon-non-cubic-CoMnGe++Co+Ni-FeGe ; workon-non-cubic-Co+Ni-Mn+Fe-P ; workon-non-cubic-CoMnAs ; workon-non-cubic-CoMnSb ; workon-non-cubic-NiCrP ; workon-non-cubic-NiMnGa ; workon-non-cubic-NiMnGe ; workon-non-cubic-Pt-Cr+Fe-Sn ; workon-non-cubic-PtMnGe++PtCrSb In the literature, there are also reports that, many samples of HHAs are seen to exhibit more than one symmetries. For example, each of NiMnAs and NiMnP are reported to possess both orthorhombic and hexagonal structures.workon-non-cubic-Co-Cr+Mn+Fe-P+As++Ni-Cr+Mn+Fe-P+As Further, in the literature, it has been found that some materials, including CoMnSbJMMM70 ; PRB74VK and PtMnAlJLM99 , exhibit site disorder, which is often associated with a larger effective unit cell. As for the magnetic properties, many of the HHAs studied in the literature are seen to be FM in nature, and it is observed that with the change in and elements, the magnetic property of the alloys changes as well.workonFMHHA
In the literature, a large number of full Heusler alloys (FHA) as well as HHAs, has been studied which shows half-metallic-like behavior but, to the best of our knowledge, only those alloys are seen to have a half-metallic-like character which possess a cubic structure. Therefore, in this work, we aim to search and screen magnetic half-metals based on half Heusler alloys: our specific interest is to probe that whether the cubic symmetry is a necessary but NOT sufficient condition for the half-metallicity in the HHA materials. It is to be further noted that, full Heusler alloys are much more extensively studied, compared to the HHAs, but none of these FHAs is seen to exhibit a structure with hexagonal symmetry. On the contrary, it is seen that many HHAs exist in different crystal symmetries, which include, hexagonal structures (with space-groups and ) as well as (face-centered) cubic ( space-group) and orthorhombic structures (with space-group ). For example, while NiMnSb is a well-known cubic HHA, the change in atom from Sb to As leads to an orthorhombic structure in the lowest energy state. There are other similar examples among the HHAs. Since many Ni-based HHAs are known to exist in phases other than the well-known cubic phase, we aim to study the reason as to why there is this difference in stability in case of various symmetries in HHAs and if there is any systematics involved. Further, in forming or Heusler alloys, atom from the p-block elements, of group IIIA, IVA and VA, are well-known in the literature. Here we probe whether atoms from group VIA are also favored in forming stable Heusler alloys.
To this end, we choose three sets of compounds, namely, Co, Ni and Pt. Choice of atom is primarily driven by the existing work in the literature, which are based mainly on Ni and also some Co and Pt-derived HHA materials. Further, the atom is chosen to be an element with high atomic magnetic moment keeping the spin-injection properties in mind. It is to be noted that we are interested in those alloys, which show both magnetism as well as half-metallicity. We study various materials in different possible space-groups to find and understand the symmetry of the phase with lowest energy for each of these materials. For this purpose, we analyze the formation energy; also partial and total density of states (DOS) in order to understand the extent of hybridization between different atoms of the alloys. Further, we try to understand the trend in similarities and differences in the magnetic and electronic properties of these three sets of materials. We specifically analyze the possibility of existence of a half-metallic-like property in these compounds in their lowest energy phases and probe if there is any one-to-one relationship between the cubic symmetry and the half-metallicity in the alloys, studied in this work. Consequently, we predict the possibility of a few new half-Heusler alloys exhibiting high spin polarization at the Fermi level (termed as henceforth) with or without having the cubic symmetry. In the next section, we discuss the methods of calculations, which are based on density functional theory. In the section followed by methodology, we present our results and discussion on the same. Finally, we summarize and conclude in the last section.
II Method
First, we discuss in detail the space-groups we have considered in our present work. We probe four different crystal symmetries, which have been reported for various half-Heusler alloys so far, namely, cubic (space-group , no. 216), orthorhombic (space-group , no. 62), as well as hexagonal structures (space-group , no. 189 and , no. 194). We have not carried out any calculations on any disordered structure in this paper due to the lack of any systematic input of structural data for the materials, studied here. In this paper, we have carried out calculations on Co, Ni and Pt-based systems; Co, Ni and Pt are taken as atom. Cr, Mn and Fe have been considered as the atom since we are interested in magnetic alloys and these atoms are known to have high atomic moments. Further, for atom, we have taken the following elements, Al, Si, P, S, Ga, Ge, As, Se, In, Sn, Sb and Te. In total, we have studied 108 different HHAs. We probe the symmetry of an alloy which has the lowest formation energy. Further, we have carried out in depth calculations of the electronic and magnetic properties of the energetically stable alloys. The half-Heusler alloys assume an ordered structure, where the and atoms are elements with d-electrons, typically transition metal (TM) atoms and atoms are elements with s,p electrons (termed as sp element).
In the lowest energy state, the most well-studied HHA NiMnSb has a structure that consists of four interpenetrating face-centered-cubic (fcc) lattices with origin at the fractional positions, (0.25, 0.25, 0.25), (0.75, 0.75, 0.75) (0.5, 0.5, 0.5), and (0.0, 0.0, 0.0). We label these sub-lattices as , , and , respectively. In structure of NiMnSb, the Ni atoms occupy the sub-lattice and the sub-lattice remain empty. Further, Mn and Sb atoms occupy the and sub-lattices, respectively.
CoMnGe is a HHA which exhibits an orthorhombic structure with symmetry. Atoms here occupy a Wyckoff position of 4c symmetry. While each of Co, Mn and Ge atoms has four equivalent atoms with fractional coordinates and as variables, the coordinate for all the four atoms is 0.25. The symmetry equivalent fractional coordinates according to the 4c point-group symmetry are as follows: ,0.25,; -+0.5,0.75,+0.5; -,0.75,-; +0.5,0.25,-+0.5.
NiMnGa assumes a hexagonal structure ( space-group) where Ni, Mn and Ga have two equivalent atoms each. Ni atom occupies sites with point-group symmetry of 2d (1/3, 2/3, 3/4 and 2/3, 1/3 and 1/4); Mn atom occupies sites with 2a symmetry (0,0,0 and 0,0,0.5); and Ga atoms are found at the sites with 2c symmetry (1/3,2/3,1/4 and 2/3,1/3,3/4).
NiFeAs is found in a hexagonal structure ( space-group). Atom Ni occupies the site with 3f point-group symmetry (,0,0; 0,,0;-,-,0); atom Fe has the preference for a site which has a 3g point-group symmetry with fractional coordinates as follows: ,0,0.5; 0,,0.5; -,-,0.5. Atom As occupies two different Wyckoff positions with 1b and 2c point-group symmetries with fractional coordinates, (0,0,0) and (1/3,2/3,0.5 and 2/3,1/3,0.5), respectively.
The equilibrium lattice constants and fractional coordinates of all these alloys have been optimized by doing full geometry optimization using Vienna Ab Initio Simulation Package (VASP)VASP which has been used in combination with the projector augmented wave (PAW) method.PAW We have interchanged the Wyckoff positions of the and atoms in case of and space-groups as well as varied the variable fractional coordinates, and in case of the latter space-group to find the structure with the lowest formation energy. and have been varied for all the three atoms , and in case of symmetry to arrive at the structure which yields the lowest formation energy among all. We report in this paper the results for the fully optimized geometries of the materials for each of the four space-groups mentioned above.
For exchange-correlation functional, generalized gradient approximation (GGA) over the local density approximation (LDA) has been used.PBE We use an optimum energy cutoff of 500 eV for the planewave basis-set. The final energies have been calculated with a mesh for which the convergence has been tested. The energy and the force tolerance for our calculations were 10 eV and 10 meV/Å, respectively. The mixing or formation energies () have been calculatedVASP for probing the energetic stability of a material, using the equation , where denotes different types of atoms present in the unit cell of the material and is the standard state (bulk) energy of the corresponding atom, .VASP These energies have been then analyzed to establish the energetic stability of the alloys in different crystal symmetries. The optimized geometries of the systems are compared with the results obtained in the literature, wherever the results are available. The detailed converged structures (fractional coordinates and lattice constants) will be reported separately.MBunpubl
For in-depth understanding of the magnetic and electronic properties, we have carried out relativistic spin-polarized all-electron calculations for the optimized structures of all the systems. These calculations have been performed using full potential linearized augmented planewave (FPLAPW) programWien2k with the generalized gradient approximation (GGA) for the exchange correlation functional.PBE For obtaining the electronic properties, the Brillouin zone (BZ) integration has been carried out using the tetrahedron method with Blöchl corrections.Wien2k An energy cut-off for the planewave expansion of about 14 Ry is typically used. The cut-off for charge density is = 14. The number of points for the self-consistent field cycles in the irreducible BZ is about 300, 600 and 2300 in case of cubic, hexagonal, and orthorhombic, respectively. The convergence criterion for the total energy is about 0.1 mRy per atom. The charge convergence is set to 0.0001.
III Results and Discussion
III.1 Analysis of Electronic Stability: Formation Energy
We study Co, Ni and Pt-based half Heusler alloys, namely, Co, Ni and Pt ( = Cr, Mn and Fe; = Al, Si, P, S, Ga, Ge, As, Se, In, Sn, Sb and Te). We calculate the formation energy of these alloys in different crystal symmetries, which include cubic (3m), orthorhombic (), as well as hexagonal ( and ) structures. As per the formation energy calculations, out of the total 108 alloys, which have been studied in this work, 25 compounds are found to be energetically unstable in any of the symmetries probed here (with close to zero or positive value of ). 35 materials have reasonably low absolute value of formation energy (below -50 kJ/mol per f.u.). Out of that, six compounds have too low a value of (lower than -10 kJ/mol per f.u.). It is to be noted here that a negative value of the formation energy obtained from the calculations indicates that at zero temperature, the compound is more stable than the bulk counterparts of the constituent elements. With a low value of formation energy, the stability of the compound is expected to be less. It is observed that out of the 108 compounds the energetically unstable ones mostly have a atom which has a large atomic number (), specifically for the Co and Ni-based alloys. Figure 1 depicts the optimized symmetry for each of the 83 energetically stable compounds, which is obtained on the basis of formation energy from our first-principles calculations. In this Figure, the symbols , , and signify , , and space-groups, respectively. From this figure, we observe that, for alloys having Co as the atom, the lowest energy structure predominantly corresponds to the orthorhombic structure. Cubic symmetry is found to be the lowest energy state, primarily for cases, which have a high element (Se, Sb and Te) as atom as well as Mn or Fe as atom. It is to be noted that for low elements as atom, the lowest energy phase is, without exception, either an orthorhombic structure or one of the hexagonal structures in all the three Co, Ni and Pt-based alloys. When atom is Ni, and atom is Mn, the alloys with a high atomic number element, namely, Se, Sb and Te as atom has cubic structure as the lowest energy state. However, for other elements, it is observed that the , and symmetries are preferred. For Pt-based systems, the situation is the same as in case of Co and Ni case: the cubic phase with high elements, Se, Sn, Sb and Te as atom, has the lowest energy. However, it is not the favored symmetry when the atom is having low . Here we point out that there are two relevant databases in the literature, where symmetries of the lowest energy state of many of the materials studied here are listed. We compare the results from these two databases here. Since Ref.heuslershome, deals with only the cubic symmetry for the half Heusler alloys, and also does not deal with the Pt-based materials, we compare only the cubic symmetry cases. We see the matching for the cases with atom with high atomic number , namely, Sn and Sb, which are expected to yield cubic ground state, is good (Figure 1). Open Quantum Materials Database (OQMD)OQMD is a more detailed database and goes beyond the Heusler alloy compounds. Out of our 108 cases, 23 systems are not listed in this database. 65 materials have been listed there against the cubic symmetry (notably, except two, all the other Pt-based systems are listed to be having cubic ground state) and overall a reasonably good matching is observed between our results and the data from this database.
To understand the relative stability of various symmetries in different compounds, the formation energies of the energetically stable 83 HHAs are shown in Tables 1 to 3. From the formation energy values we find that, many of the 83 materials are likely to exist in more than one crystal symmetry since the formation energies of these different symmetries are within a few meV per formula unit (f.u.) of each other. In Tables 1, 2 and 3, we highlight (in bold) the entries corresponding to the lowest formation energy. The experimentally available structures are also presented in these tables against the respective compound. We find that the predicted symmetries with the lowest energy for different materials, by and large, match with the literature, except for a very few materials.
Results on Cubic case – From Figure 1, we observe that group VIA seems the most favorable atom for formation of the HHAs in the cubic symmetry with structure (space-group ). It is further observed that for larger atoms of other groups as well (for example, In, Sn, and Sb) the cubic phase is more stable compared to other symmetries. Among materials studied here, 68 compounds seem to have energetically stable cubic phase, ground state or not. Out of that maximum (30) is Pt-based compounds. From theoretical study, many of these cubic compounds are reported to show half-metallic-like character in the cubic structure.PRB95JM However, only a few among these have been experimentally synthesized and found to possess cubic structure.workon-non-cubic-CoMnSb ; workonNiMnSb ; JMMM38 ; workon-non-cubic-Pt-Cr+Fe-Sn
Results on Hexagonal symmetry – In total 69 compounds out of 108 have negative formation energy in the case of hexagonal Ni2In type structure (with space-group ). Compared to the Co atom, with Ni and Pt atoms at the site, more number of alloys seems to be having negative formation energy. Experimentally, CoFeGe is reported to have hexagonal structure. However, from our calculations the lowest energy state of CoFeGe is found to be orthorhombic; but, the formation energy in the hexagonal () is close to that of the orthorhombic structure (Table 1). CoMnSi, CoMnGe and NiMnSi compounds are reported to exist in both hexagonal () and orthorhombic () structure. Our present calculation shows that the lowest energy state structure of these compounds is orthorhombic.
Results on Hexagonal symmetry – From our calculations, 68 compounds are found to possess a negative formation energy for the hexagonal structure with a space-group of . Out of these compounds quite a few are experimentally synthesized and are indeed found to have hexagonal structure.workon-non-cubic-Co-Cr+Mn+Fe-P+As++Ni-Cr+Mn+Fe-P+As ; workon-non-cubic-Co+Ni-Cr+Mn+Fe-P++Co+Ni-Cr+Mn-As ; workon-non-cubic-Co+Ni-Mn+Fe-P However, there are few small differences. For example, NiCrSi is experimentally observed to have an orthohrombic structure. But from our calculation the lowest energy state is found to be hexagonal , though it is to be noted here that the formation energy of orthorhombic structure is very close to that of the hexagonal structure. The energy difference is 3.42 kJ/mol (34.2 meV per f.u.) which is of the order of the thermal energy. Experimentally, NiMnGe is found to possess orthorhombic and hexagonal () structures. But from our calculation, the obtained lowest energy state is the orthorhombic structure. We see from Table 2 that, the formation energies for the orthorhombic and the two hexagonal structures are reasonably close to each other. Similar is the case for CoFeAs. It is seen to have a hexagonal structure from literature. But from our calculations, it is observed that both the and the hexagonal phase have very close value of , and the former is slightly more stable than the other (Table 1).
Results on Orthorhombic phase – From our calculations the compounds, which are found to be in orthorhombic, NiTiSn type structure with space-group , are primarily Co-based compounds. In total 70 alloys have energetically stable structure. It is worth-noting that for most of the NiMn structures, with a atom with a low value, the ground state is found to be energetically very close to the orthorhombic phase. The symmetry of compounds at the lowest energy state matches with the reports in the literature, barring a few exceptions. While CoCrAs, CoMnGe and NiCrAs are found to exhibit a hexagonal symmetry, our calculations yield an orthorhombic lowest energy state for these alloys. However, it is to be noted that from Table 1 and 2, we observe that the formation energies of both these structures are very close (difference being 3 to 5 kJ/mol per f.u.). Another exception is the case of CoMnAs. It is found to possess orthorhombic phase as ground state but from experiment it is found to have space-group. The energy difference, however, in this case, is large between these two phases (Table 1). For many compounds it is found that, the orthorhombic phase has a formation energy which is very close (within 5 meV per f.u.) to the of one of the hexagonal phases. Hence, from our detailed analysis of values, it can be inferred that the orthorhombic structure is the most common symmetry among the Co-based materials. There are quite a few of the Ni and Pt-based materials for which also this structure has the lowest energy. As discussed above, in many cases two or more phases are close in energy. Hence, it is likely that samples of the non-ground state phases of these materials may actually form under certain experimental conditions.
III.2 Geometry Analysis: Lowest Energy State versus Other States
The lattice parameters for all the energetically stable materials are given in Tables 1 to 3. The cubic phase has a relatively open structure. Hence, a cubic phase is found to be the lowest energy state for materials with atoms with larger atomic radii and larger values (of Pt and some of the atoms).
There are about twelve systems where the values of two phases are very close and these values are within 1 kJ/mol per f.u. To understand this small difference in the , we perform a detailed geometry analysis. Table 4 gives relevant data for some typical materials, where in a few cases there is an excellent matching of the between two symmetries and where there is no matching. In this table, along with the density values and the bondlength between two atoms (), we have also noted if the simulated X-ray diffraction (XRD) patternVASP of the phase matches with the same of the ground state structure or not. We present the data of all the symmetries for each material where first, second, third and fourth rows correspond to the ground state () and the three other phases, having difference in formation energy with the ground state in an increasing order: these are described as +1, +2 and +3 states. For PtCrSn and PtMnIn it is observed from Table 3 that, is the ground state. However, it is seen that the respective phases, that is the +1 phase in these two systems, possess a energy which is very close to the ground state (within 0.01 kJ/mol/f.u.). Similarly, PtFeIn has a structure as the lowest energy state and structure also has a very similar value for the formation energy (Table 3). From Table 4 it is clear that the XRD patterns of these two symmetries for the three above-mentioned materials are expected to be close. Additionally, the densities of these two phases for PtCrSn, PtMnIn and PtFeIn are close to each other. When we compare the bondlength between the atoms -, - and - for the two symmetries for these three materials, we note that the values vary maximum by only 0.03 Å. Since the density, bondlengths and the simulated XRD patters match so well, it is clear that the internal local geometries of each atom and subsequently the bonding nature in the two phases with two different symmetries for each of these three materials are the same. It is also clear from Tables 1 to 4 that the other two symmetries ( and ) are not only energetically farther from the ground state, but these are also different from the geometric point of view. In case of NiCrGa (Table 2), and PtCrIn (Table 3) also similar results are obtained. While these two symmetries ( and ) in NiCrGa show good overall matching, in case of PtCrIn, the XRD patterns are found to be not quite close.
Further, it is interesting to probe PtMnSn and PtMnSb for the following reason. It is observed that the ground state symmetry is cubic in both the cases. When the data for these two materials from Table 4 are analyzed it is found that, while the geometric data of the ground state symmetry does not match with those of any of the other three symmetries, all the data from second and third row (for phases and ) match very well. Table 3 lists the respective formation energies for these two materials and we observe that these data are indeed consistent with this observation. Subsequently, the XRD patterns of these materials for the two above-mentioned symmetries are seen to resemble each other. Next we discuss a few cases, where the value of one symmetry is only somewhat close to the ground state. We take the example of NiMnAs. The ground state has a (-73.01 kJ/mol per f.u.) and density values (7.75 Mg/m3); the symmetry has somewhat close values for these two quantities (-71.68 kJ/mol per f.u. and 7.67 Mg/m3, respectively). Consequently, the geometric data including simulated XRD patterns of the two phases also exhibit not a good matching with each other. Similar is the case for materials, for example, NiMnAl, CoCrGe, PtMnAl and PtMnGe.
III.3 Analysis of Total and Partial Moment
In this subsection, we discuss the results on the magnetic properties of the materials, studied here. The calculations are carried out with a magnetic configuration for all the materials. Since Co has a significant moment, there is a possibility that a ferrimagnetic (moments of Co and atom aligned anti-parallel to each other) configuration may be likely. After convergence we get, in a few cases a ferrimagnetic and in most of the cases a FM configuration as observed in the literature.PRB95JM ; PRB74VK We present in Tables 5 to 7, the total, partial moments, the total number of valence electrons, and the of all the materials, wherever possible. The results of the cubic case and the lowest energy state obtained from our calculations are listed in these tables. When cubic is the symmetry for the lowest energy state, the explicit entries corresponding to the cubic case (on the left side of the Tables 5 to 7) are left empty. Values corresponding to energetically unstable cases have also been put (in italics) to see whether any trends which are found for the stable cases are followed by these or not.
Slater-Pauling rule, and Integer Moment versus Half-metallicity in Cubic case – It is observed in the literature that many Co-based Heusler alloys, specifically the half-metallic ones, follow the Slater-Pauling rule.JMMM423TR ; JPD40HCK ; PRB66IG As a consequence of this rule, an almost linear variation of the magnetic moment with the atomic number of the atoms for the cubic case of the Co-based FHA materials is observed. In this work also we expect that a linearly increasing trend of the total moment as a function of the value of the atom will be observed in the cubic cases of Co. Figure 2 shows the CoMn cases. The cases with positive are also plotted for overall comparison. We find that the linear trend is not quite followed when the of the compound is positive. However, the total moment of the energetically stable alloys shows this linear trend as is clear from both Figure 2 and Table 5. These findings are true for CoCr systems as well. We further observe from Figure 2 that though like the total moment, the partial moments, mainly that of atom also show an increasing trend, there is a change of slope in both the cases of the and atoms, as is true for stable CoCr systems also. It is to be noted that none of these trends are followed in case of CoFe. Figure 2 exhibits a few Ni and all Pt-based cases as well. In case of Ni and Pt-based systems, in general, none of these above-mentioned trends is observed. The moments of the NiFe alloys are seen to all together deviate from the trend. On the contrary, NiCr and NiMn cases with atoms from group IVA, VA and VIA tend to follow the Slater-Pauling rule, when the is negative and atom has a lower atomic number. As the group of the atoms changes, from IVA to VA to VIA, the total moment increases (by value 1), and partial moment of both Ni and atoms increases. This observation has been discussed again in the next subsection in terms of the DOS of the up and down spin electrons. As the group of the atom remains the same and the period changes, the total moment remains the same, but moment of Ni and atoms decreases and increases, respectively. For the Pt-based systems, the Slater-Pauling rule of linear increase of total moments is not obeyed (Figure 2). Further, it has been observed that for the cubic cases, in many of the materials (maximum for the Co-based systems), the moment is integral in nature. It is expected that typically an integral moment leads to a half-metallic system in cubic phase. It will be discussed in detail in the next subsection after analyzing the total density of states of the up and down spin electrons, for both cubic and non-cubic systems.
Partial Moments in the Cubic case – Here we concentrate on the cubic phases of the materials. It is observed from the partial moments that for some of the cases the final configuration has turned out to be ferrimagnetic. In the literature, it has been discussedPRB95JM that there is a dependence of the long-range magnetic configuration on the number of valence electrons (NVE). While systems with NVE=18 (case a) show anti-ferromagnetism, cases with NVE=19 and 20 (case b) exhibit ferrimagnetism. On the contrary, NVE=21 and 22 (case c) lead to ferromagnetism. The atom carries the maximum moment in all these cases discussed here. The moment on the atom is almost equal in magnitude and opposite in direction in case a. In case b, moment on is smaller in magnitude but oriented in an anti-parallel arrangement with respect to moment of atom. On the contrary, case c deals with a long range FM configuration, where both and moments, though may be unequal, orient along the same direction. By analyzing our results (Tables 5, 6 and 7) on the partial moments of the energetically stable cubic phase, we find that there are only a few exceptions. While Co and Ni-based systems generally follow the trend, maximum deviation is observed in case of Pt-based systems. We have found that for NVE=23 also, the results follow the trend as in case c.
Total and Partial moments in the Ground states – Here we concentrate on the total and partial moments of the phases with the lowest energy. Few of the cases exhibit a close-to-integral total moment (CoMnP, CoFeP, CoFeS, NiCrAs, NiFeS, PtCrP, PtMnSi, PtFeIn all in symmetry as well as PtMnIn in symmetry). Like a cubic case, there may be a possibility of half-metallicity in these non-cubic cases. A detailed analysis of DOS is warranted for the validation of the same (see next subsection). We analyze here the partial moments on the different atoms, present in the system. It is seen that in most of the cases the moments on the and atoms are anti-parellel to each other (Tables 5 to 7). However, the moments on the latter atoms are much smaller compared to the earlier ones as is observed in the cubic phase as well. Unlike the cubic case, no increasing trend in total moment as a function of of the atom is observed for the lowest energy state. No trend is observed in the values of the total moments when the cubic and the states of any of the materials is compared. In majority of the cases the moments on the and atoms are found to be parallel to each other, including the Co-based compounds.
Spin Polarization at the Fermi Level – Next we discuss about the extent of spin polarization at the Fermi level () of various materials in cubic versus the ground state. It is observed that except few of the =Fe atom cases, Co materials possess high SP in the cubic case. For Ni, only with the exception of NiMnSn and NiMnTe, all the cubic cases exhibit high SP. On the other hand, for Pt, for the cubic cases seems to be below 50% for many of the alloys. Many, but not all, of the cubic structures of different materials exhibit a 100% . However, this is not the case with the lowest energy structure of any of the materials, which are studied in this paper. In the lowest energy case, we observe that only a few materials, with or without integral total moment, possess high , which is above 65%. While there are 6 of these, but none has a 100% . Out of these 6 cases, in ground state NiCrAs () with a of 69.6 % and PtCrP () with a of 66.4 % have a total integral moment of 2.99 and 3 , respectively. Further, for PtFeGe () has a moment of 1.99 and value of 71.9 %. However, while in the ground state, CoCrP (), NiFeSi (), NiFeP (), and PtMnP () have comparable values of 76.3 %, 71.2 %, 69.7 % and 84.2 %, respectively, the corresponding total moments are 1.93, 1.72, 1.08 and 2.12 , respectively.
III.4 Analysis of Electronic Structure
Density of States of the Cubic Phase – For overall comparison of the trends, we plot in Figure 3, 4 and 5 the up and down DOS of the cubic and the lowest energy state of all the materials, including the ones with positive . It is observed that the trends which are followed in case of the energetically stable ones are not strictly followed in case of the ones with positive . The following trend is observed from Figure 3 for Co alloys. As the number of valence electrons of the atom increases in a period, for example, Al to Si to P to S, the valence band width (VBW) is seen to increase systematically. Further, specifically for all the other cases, except a atom from group VIA, the DOS shifts away from Fermi level, leading to a higher binding energy of the system. The DOS at and very close to Fermi level also changes. In case of S, Se and Te, these systematics are not consistently followed throughout. Furthermore, it is seen that when the atom is from group IIIA, all the alloys of type Co are found to be energetically unstable. As the value of atom increases in a group, for other groups, for example, Si to Ge to Sn, i.e. atoms with same NVE, not only the VBW, but also the DOS at the Fermi level remains similar. When the atom changes, namely, Co atom is replaced by Ni and Pt, the shifting of the weight of the DOS curve towards lower energy (more binding energy) is clearly evident from Figures 4 and 5. The overall trends, near the Fermi level, as discussed above, are found to be the same. However, it is observed that while for =Ni, most of the alloys are predicted to be energetically unstable, when the atom is from group IIIA. For Pt all the alloys seem to have negative and it is further to be noted that the weight of the DOS shifts towards lower energy making the systems more bound in comparison to = Co or Ni, as is evident from the formation energies given in Tables 1 to 3 as well. As to why there is dip in the total moment (Figure 2), when the atom is mainly an Mn atom and atom is from group IVA, can be understood by analyzing the respective DOS curves. The up and down DOS in this case are seen to be more compensated leading to a lower moment when compared to the cases where atoms are from other groups (Figures 3 to 5). As the value of atom increases and the and atoms remain the same, the energy is lowered as of atom increases, since the NVE of the system increases. However, it has been noted that, largely, the down spin DOS is more involved in all these, compared to the DOS of the up spin.
Comparison of DOS of Cubic versus Lowest Energy State – Figure 6, 7 and 8 give the total and partial DOS of the cubic and ground state of few materials. We have chosen different materials with various symmetries: while Figure 6 and 7 exhibit the DOS of and symmetries, respectively, DOS of few materials for which space-group is energetically the most favorable, is given in Figure 8. It is observed that as in these cases, and also in many other cases, there is a shift of weight of DOS of the ground state towards lower energy compared to the cubic state. Further, after critical analysis of DOS of ground versus the cubic state, the following few general points become note-worthy and relevant with regard to the non-cubic ground state symmetry of many materials (Tables 1 to 3). Mainly in the down spin DOS, a significant hybridization is noted between the and atoms, specifically, very close to the Fermi level.MBunpubl A double-peak structure between the DOS of these two atoms is prominent in most of the cases. Further, hybridization between the and atoms and sometimes among all the three atoms is also observed. More than often this is found in the lower energy ranges (away from Fermi level) and also typically for up spin DOS. While cases with both Mn and Cr as atoms show similar results, cases with Fe as atom are slightly different: for example, the overlapping behavior with and/or atoms is better for up spin DOS and at lower energy compared to the vicinity of the Fermi level. These observations may explain the following: though, in the literature there is a lot of theoretical study on the cubic phases and half-metallicity in case of HHAs, experimentally many HHAs have been shown to prefer a lower symmetric structure and discussion about the HM-like behavior in a non-cubic case is generally missing from the literature.
DOS of Two Symmetries with Close – Figure 9 gives the density of states for a few materials, for which two symmetries yield close to very close and also geometry (Tables 1 to 4). As is evident from Table 3, for the material PtCrSn, two symetries ( and ) possess the same within our calculational accuracy. The lowest panel in Figure 9 gives the support for the same from the electronic structure calculations. The total as well as partial DOS for this material in these two symmetries have an excellent matching. Similar is the case for PtMnIn and PtFeIn where the values for these two phases are also very close.MBunpubl Further, two materials PtMnSn and PtMnSb have a cubic phase as a lowest energy state. However, in the +1 (symmetry with higher energy than the ground state) and +2 (symmetry with higher energy than the +1 state) cases, PtMnSn has and , respectively, while PtMnSb has and , respectively. These two phases also have very similar formation energy. In Figure 9, we show the total and partial DOS for these +1 and +2 cases for PtMnSn and we observe that the DOS for these two cases are again matching very well. Similar is the case for PtMnSb.MBunpubl As is evident from Tables 1 to 4 and Figure 9, the results of structural and electronic structure calculations are consistent for all these five materials mentioned here. On the contrary, when the for one phase is slightly higer than the other (as in case of CeFeAs and NiMnAl), the peak positions and intensities of different peaks are not so alike as discussed above. Though in these two cases the is different by only about 0.5 kJ/mol, from upper two panels it is seen that the matching of DOS of the two phases is not so excellent. The matching is slightly better in case of NiMnAl where the two symmetries involved are and . This has been a general observation throughout. If symmetry is found to be close to symmetry, the matching of the geometry and electronic structure are very good. However, regarding the matching between and symmetries, geometrical and electronic structures are seen to match only reasonably for these two phases even if energetically these phases are close to each other.MBunpubl From literature on experimental studies of HHAs, it is observed that a very few cases exist where a material is reported to be in two different symmetries. Pertinent examples of such cases are NiMnAs, NiMnP, CoMnGe and NiMnGe. Figure 10 shows the DOS of the two experimentally reported phases for these materials. It is observed that both partial and total DOS are quite close for these two phases for all these materials. Since the difference between the two values are more in case of CoMnGe, somewhat significant differences in total and partial DOS are visible from Figure 10.
DOS of Non-cubic HM-like States – Finally, we show the plot of density of states for a few materials which are likely to exhibit a HM-like behavior depending on the values of total (integral) moment in their non-cubic ground state. Figure 11 exhibits the DOS of CoMnP, CoFeP, NiCrAs and PtCrP which show a gap or a pseudo-gap with a very low DOS at the Fermi level, for one of the spin channels, for both the ground state and the cubic phase. The cases of NiCrAs and PtCrP in the lowest energy state ( phase) are reasonably clear. A very small density of states at the Fermi level is observed for the down spin channel of both the materials. However, the DOS at the up spin channel is also somewhat small for both the cases. This results in an effective spin polarization (at the Fermi level) of about 70 and 66% for NiCrAs and PtCrP in the ground state, respectively. Contrary to these cases, both CoMnP and CoFeP will probably behave as a bad semi-metal rather than a half-metal since there are small densities of states at both the spin channels, up DOS being slightly larger in intensity than the down DOS. We note from Tables 5 to 7 that, these four materials possess total moments which are very close to integers. Total moment of 3, 2, 2 and 3 are observed for CoMnP, CoFeP, NiCrAs and PtCrP, respectively. There are few other materials, for which also, in the non-cubic case, the total moment is very close to an integer. These cases include NiFeS, PtMnSi, PtFeIn - having the (See Tables 5 to 7), and also PtMnIn - having the space-group, respectively. In these cases also, our calculations reveal the appearence of a pseudo-gap at the down spin channel.MBunpubl However, the DOS at the Fermi level for the up spin channel, though is higher compared to the DOS for the down spin electron, the absolute value of the DOS for down spin electron is not negligible, as is observed in the case of NiCrAs and PtCrP. Further, we consider the cases of some other materials with high but with total moment not so close to an integer value. It has been observed that out of these materials, in the ground state, CoCrP and NiFeSi have , and NiFeP and PtMnP have symmetries; and as discussed above, these have total moment of 1.93, 1.72, 1.08 and 2.12 , respectively. The corresponding has been observed to be high and comparable to those of NiCrAs and PtCrP. Therefore, we observe that, though there is no rigorous one-to-one relationship among the cubic symmetry, integral total moment and high , but a strictly half-metallic behavior (with exactly 100% ) and integral total moment are found to be associated only with the cubic symmetry. Hence, by combined analysis of magnetic and electronic structure calculations, CoCrP, NiCrAs, NiFeSi, NiFeP, PtCrP and PtMnP alloys are predicted to be non-cubic half Heusler alloys with significantly high . However, the point to be noted here is that a high does not necessarily indicate a possibility of semiconducting behavior along one spin channel, and in turn, a possible application of a material as an appropriate spin-injector material. Hence we predict from our present density of states calculations that NiCrAs and PtCrP are the only two non-cubic materials which in their phase may have a potential in this regard and also these are magnetic in nature. This observation awaits the experimental validation.
IV Conclusion
In this paper geometric, electronic, and magnetic properties of Ni, Co and Pt-based half Heusler alloys, namely, Ni, Co and Pt ( = Cr, Mn and Fe; = Al, Si, P, S, Ga, Ge, As, Se, In, Sn, Sb and Te) have been calculated in detail using first principles calculations based on density functional theory. Quite a few of these materials with a atom from group IIIA, IVA and VA have already been experimentally and/or theoretically found in various different symmetries. In this work, we probe the stability of all the above-mentioned alloys in different crystal symmetries, reported in the literature. These structures include, the most common (face-centered) cubic phase (space-group ), and also orthorhombic (space-group ), as well as hexagonal (space-groups and ) phases. We find from our calculations of formation energy that along with alloys with elements from group IIIA, IVA and VA, alloys with elements from group VIA are also, by and large, energetically stable. It has also been observed that high elements as the atom lead to stabilized phases in case of the Pt-based compounds. On the contrary, it is not so in the case of Co and Ni-based materials.
In literature half-metallicity in many half and full Heusler alloys have been shown to exist which is typically associated with a cubic symmetry. We note from the results of the magnetic properties calculations, that there is a possibility of existence of some novel non-cubic half-metallic-like half Heusler alloys, as these possess total integer moments. Therefore, to discuss the relative stabilities of different symmetries in order to search for the respective lowest energy state for all the materials as well as to ascertain whether a material is half-metallic or not, we analyze the partial and total density of states. Based on the results of the magnetic and electronic properties, (i) we show that for a material depending on the hybridization between different atoms a particular symmetry is more stable compared to the cubic or other phases; (ii) we observe that there is no rigorous one-to-one relationship between the cubic symmetry and high spin polarization at the Fermi level; (iii) it is found that a strictly half-metallic behavior (with 100% spin polarization) is associated only with the cubic symmetry; (iv) along with a few new cubic half-metallic alloys, we predict the possibility of existence of a few novel non-cubic alloys with significantly low DOS in one of the spin channels and high spin polarization at the Fermi level.
V Acknowledgement
Authors thank P. A. Naik, T. Ganguli and Arup Banerjee for facilities and constant encouragement throughout the work. The scientific computing group of computer centre, RRCAT, Indore and P. Thander are thanked for help in installing and support in running the codes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1(1) I. Zutic, J. Fabian, S. Das Sharma, Rev. Mod. Phys. 76 , 323 (2004); S.A. Wolf, D.D. Awschalom, R.A. Buhrman, J.M. Daughton, S. von Molnar, M.L. Roukes, A.Y. Chtchelkanova, D.M. Treger, Science, 294 , 1488 (2001).
- 2(2) R.A.de Groot, F.M. Mueller, P.G. van Engen, K.H.J. Buschow, Phys. Rev.Lett., 50 , 2024 (1983); R.A. de Groot, K.H.J.Buschow, J. Magn. and Magn. Mater., 54–57 , 1377 (1986) ; I. Galanakis, P. H. Dederichs and N. Papanikolaou P Hys. Rev. B 66 , 134428 (2002).
- 3(3) J. Ma, V. I. Hegde, K. Munira, Y. Xie, S. Keshavarz, D. T. Mildebrath, C. Wolverton, A. W. Ghosh, W. H. Butler, Phys. Rev. B, 95 , 024411 (2017) and the references therein.
- 4(4) Tufan Roy, Dhanshree Pandey, Aparna Chakrabarti, Phys. Rev. B, 93 , 184102 (2016) and references therein.
- 5(5) Tufan Roy, Aparna Chakrabarti, J. Magn. Magn. Mater., 423 , 396 (2017).
- 6(6) P. G. van Engen, K. H. J. Buschow, R. Jongebreur, M. Erman, Appl. Phys. Lett., 42 , 202 (1983); P. J. Webster, K. R. A. Ziebeck, Alloys and Compounds of d-elements with Main Group Elements, Springer, Berlin, Germany, 75 (1988); K. R. A. Ziebeck, K. U. Neumann, Magnetic Properties of Metals, Springer, Berlin, Germany, 64 (2001); H.C. Kandpal, G.H. Fecher, C. Felser, J. Phys. D: Appl. Phys., 40 , 1507 (2007); H. Zenasni, H.I. Faraoun, C Esling, J. Magn. Magn. Mater., 333 , 162 (2013)
- 7(7) T. Block, M. J. Carey, B. A. Gurney, O. Jespen, Phys. Rev. B, 70 , 205114 (2004); S. Gardelis, J. Androulakis, P. Migiakis, J. Giapintzakis, S. K. Clowes, Y.Bugoslavsky, W. R. Branford, Y. Miyoshi, L. F. Cohen, J. Appl. Phys., 95 , 8063 (2004); S. K. Ren, J. Gao, X. L. Jiang, G. B. Ji, W. Q. Zou, F. M. Zhang, Y. W. Du, J. Alloy Comp., 384 , 22 (2004); S. K. Ren, W. Q. Zou, J. Gao, X. L. Jiang, F. M. Zhang, Y. W. Du, J. Magn. Magn. Mater., 288 , 276 (2005); Z. Wen, T. Kubota, T. Yamamo
- 8(8) E. L. Habbak, R. Shabara, S. H. Aly and S. Yehia, Physica B : Cond. Mater., 494 , 63 (2016); V. A. Dinh, K. Sato and H. K.Yoshida, IEEE Trans. Magnetics, 45 , 2663 (2009); S. Y. Lin, X. B. Yang, Y. J. Zhao, J. Magn. Magn. Mater., 350 , 119 (2014); Y. Wu, B Wu, Z. Wei, Z. Zhou, C. Zhao, Y. Xiong, S. Tou, S. Yang, B. Zhou, Y. Shao, Intermetallics, 53 , 26 (2014); M.P. Ghimirea, Sandeep, T.P. Sinha, R.K. Thapa, J. Alloys and Comp., 509 , 9742 (2011); L. Offernes, P. Ravindran, A. Kjeksh