AFLOW$\pi$: A minimalist approach to high-throughput ab initio calculations including the generation of tight-binding hamiltonians
A. R. Supka, T. E. Lyons, L. Liyanage, P. D'Amico, R. Al Rahal Al, Orabi, S. Mahatara, P. Gopal, C. Toher, D. Ceresoli, A. Calzolari, S., Curtarolo, M. Buongiorno Nardelli, and M. Fornari

TL;DR
AFLOWπ is a streamlined framework for high-throughput ab initio calculations that automatically generates tight-binding Hamiltonians and simplifies complex property computations.
Contribution
It introduces AFLOWπ, a minimalist tool that automates tight-binding Hamiltonian generation and integrates various advanced material property calculations.
Findings
Automated generation of tight-binding Hamiltonians from first principles.
Successful computation of optical, elastic, and vibrational properties for various materials.
Simplified workflow for complex electronic and phononic property calculations.
Abstract
Tight-binding models provide a conceptually transparent and computationally efficient method to represent the electronic properties of materials. With AFLOW we introduce a framework for high-throughput first principles calculations that automatically generates tight-binding hamiltonians without any additional input. Several additional features are included in AFLOW with the intent to simplify the self-consistent calculation of Hubbard U corrections, the calculations of phonon dispersions, elastic properties, complex dielectric constants, and electronic transport coefficients. As examples we show how to compute the optical properties of layered nitrides in the N family, and the elastic and vibrational properties of binary halides with CsCl and NaCl structure.
Click any figure to enlarge with its caption.
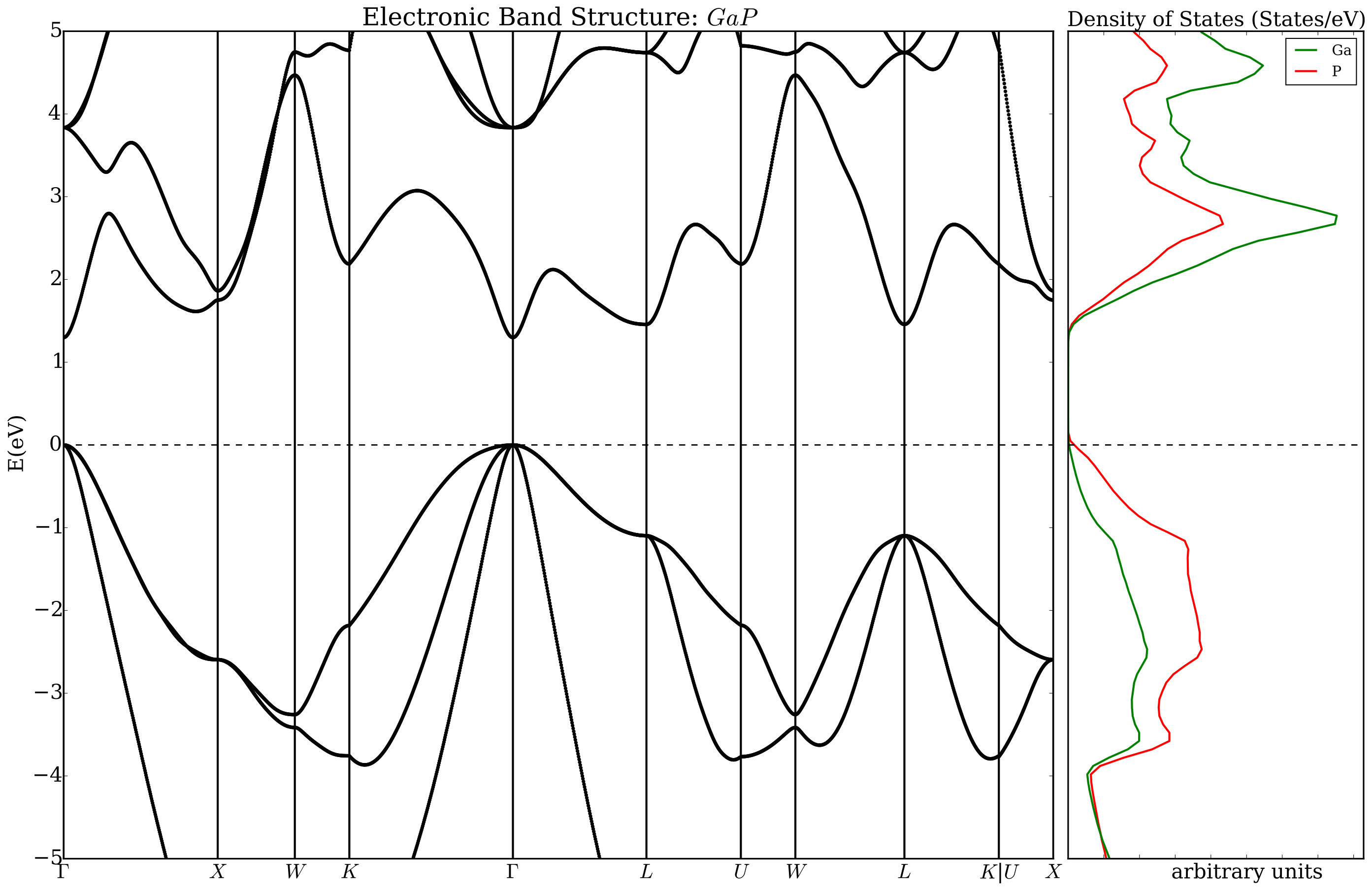 Figure 1
Figure 1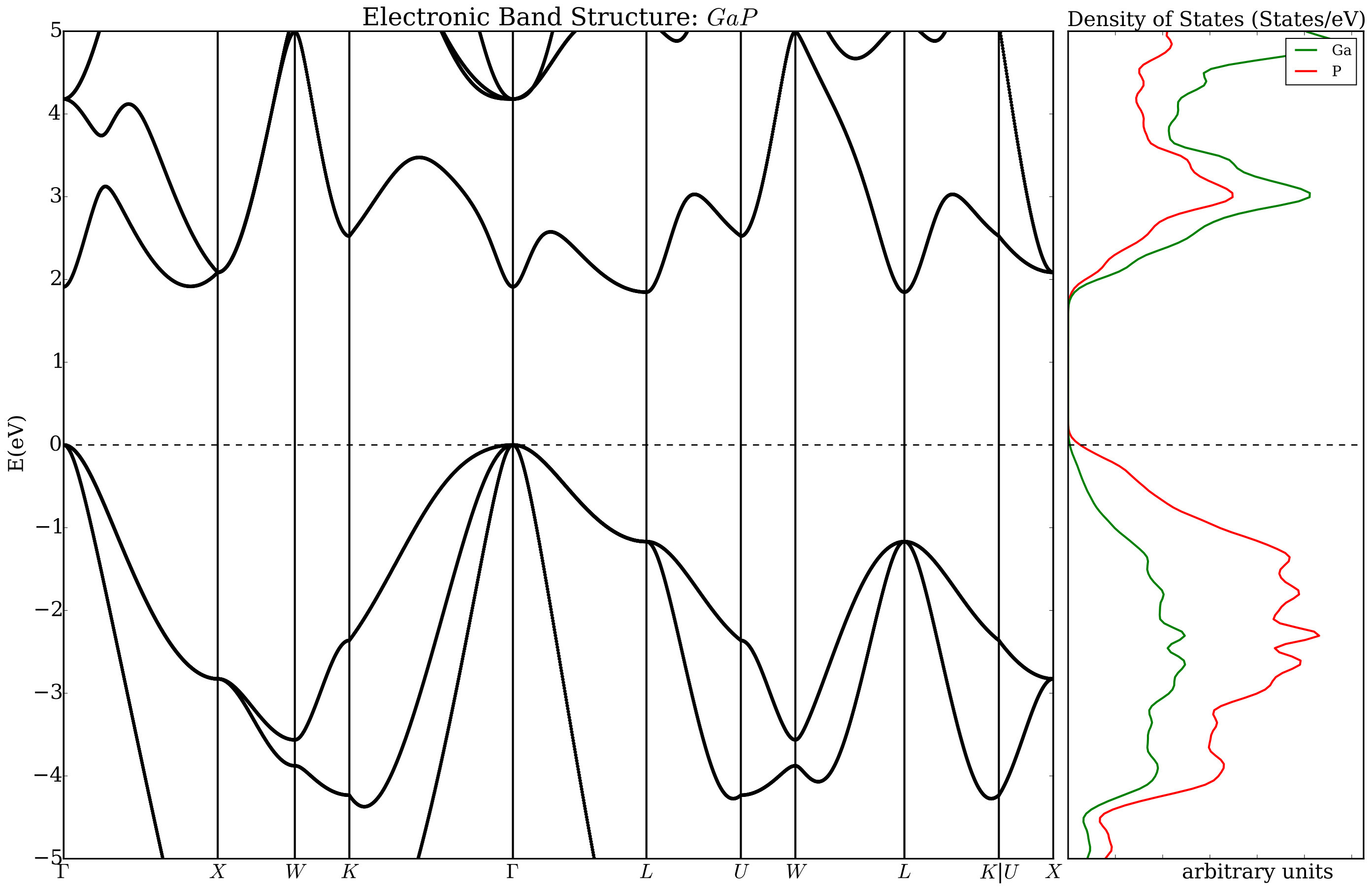 Figure 2
Figure 2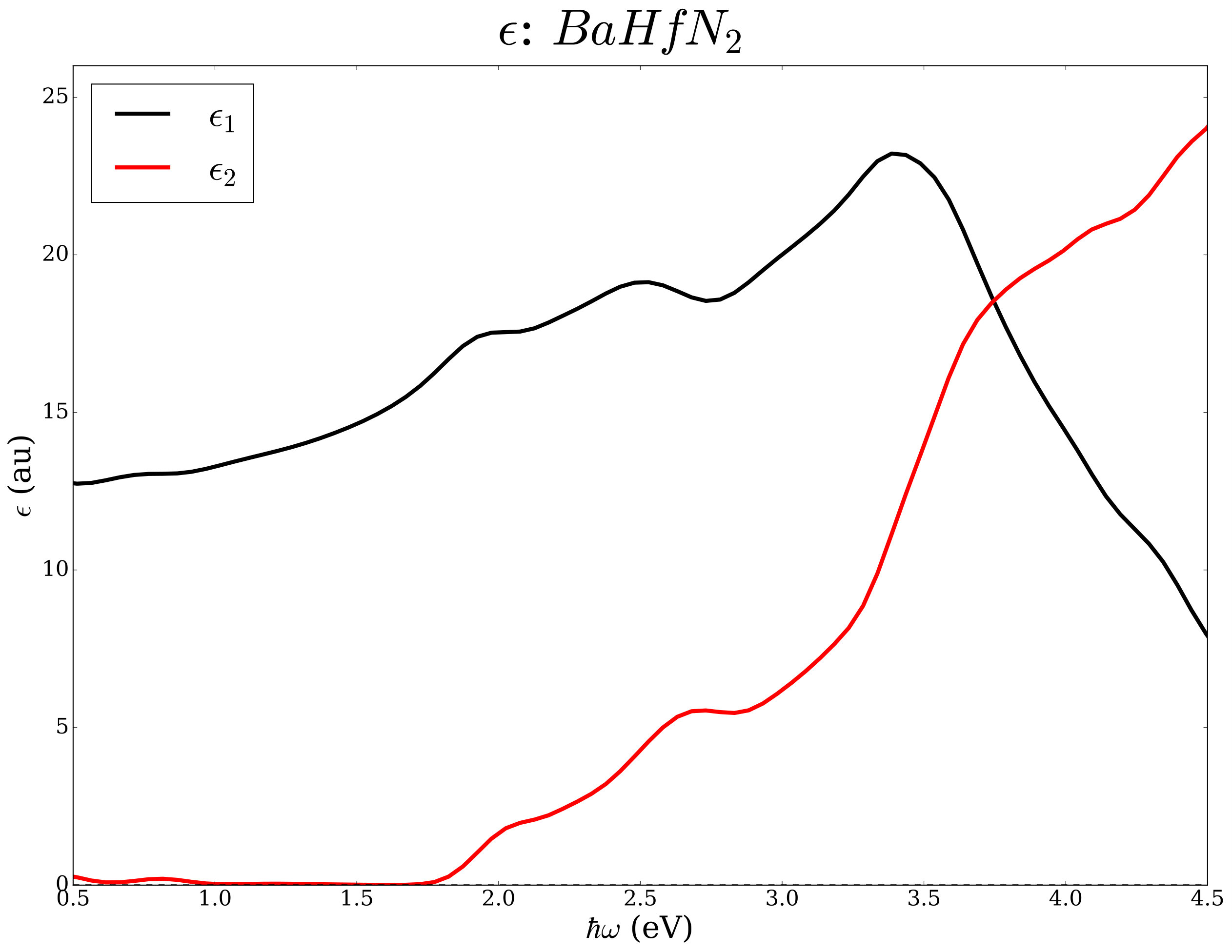 Figure 3
Figure 3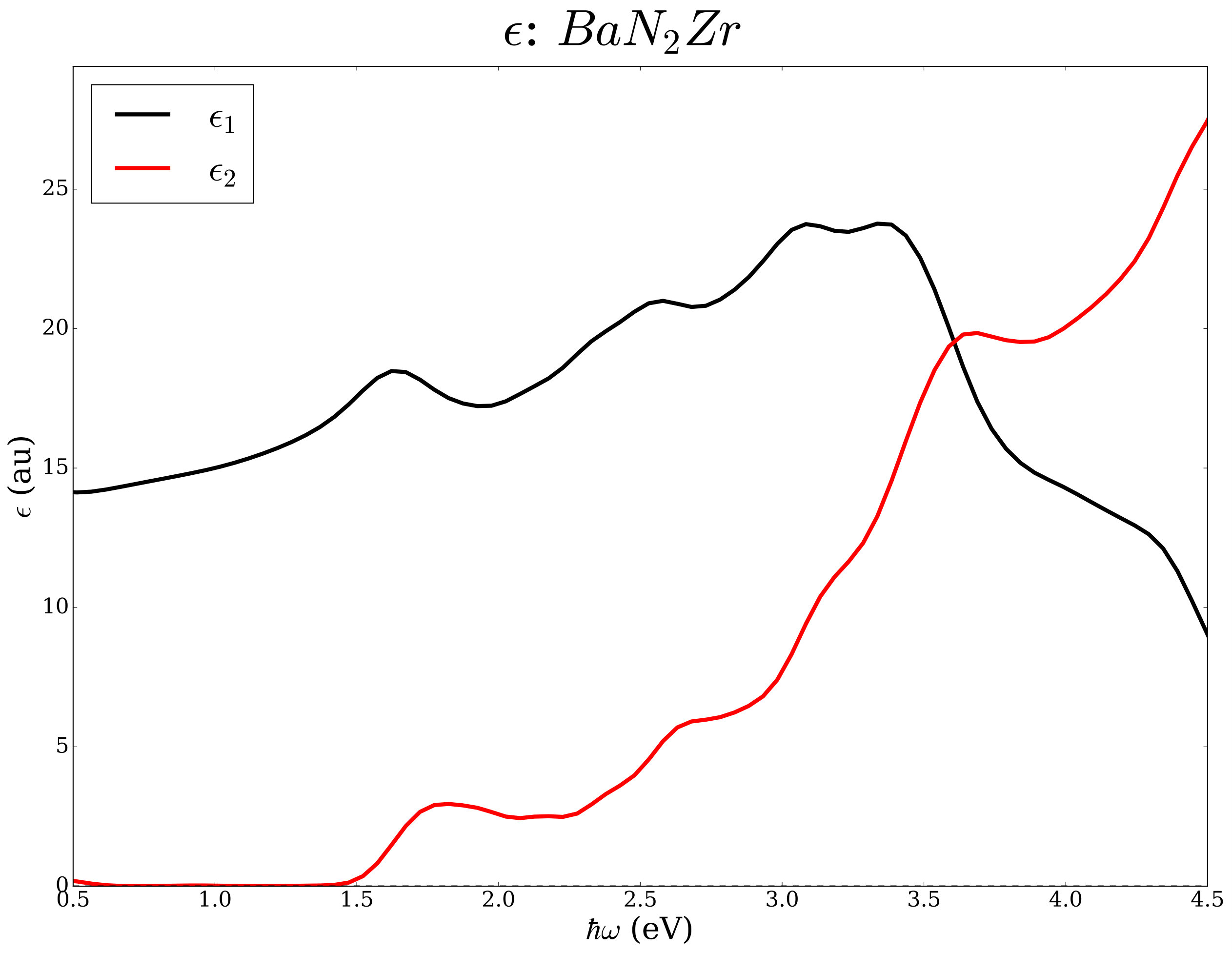 Figure 4
Figure 4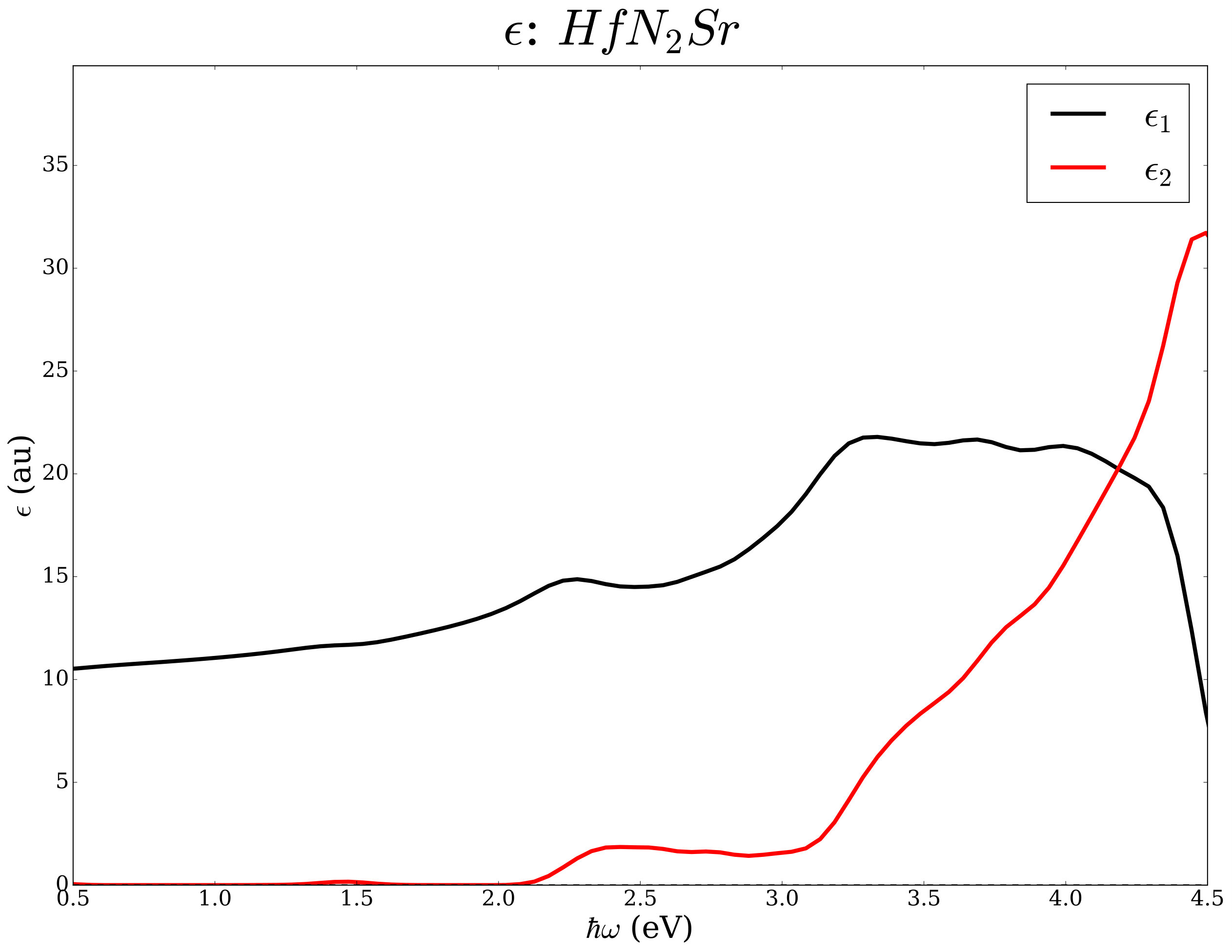 Figure 5
Figure 5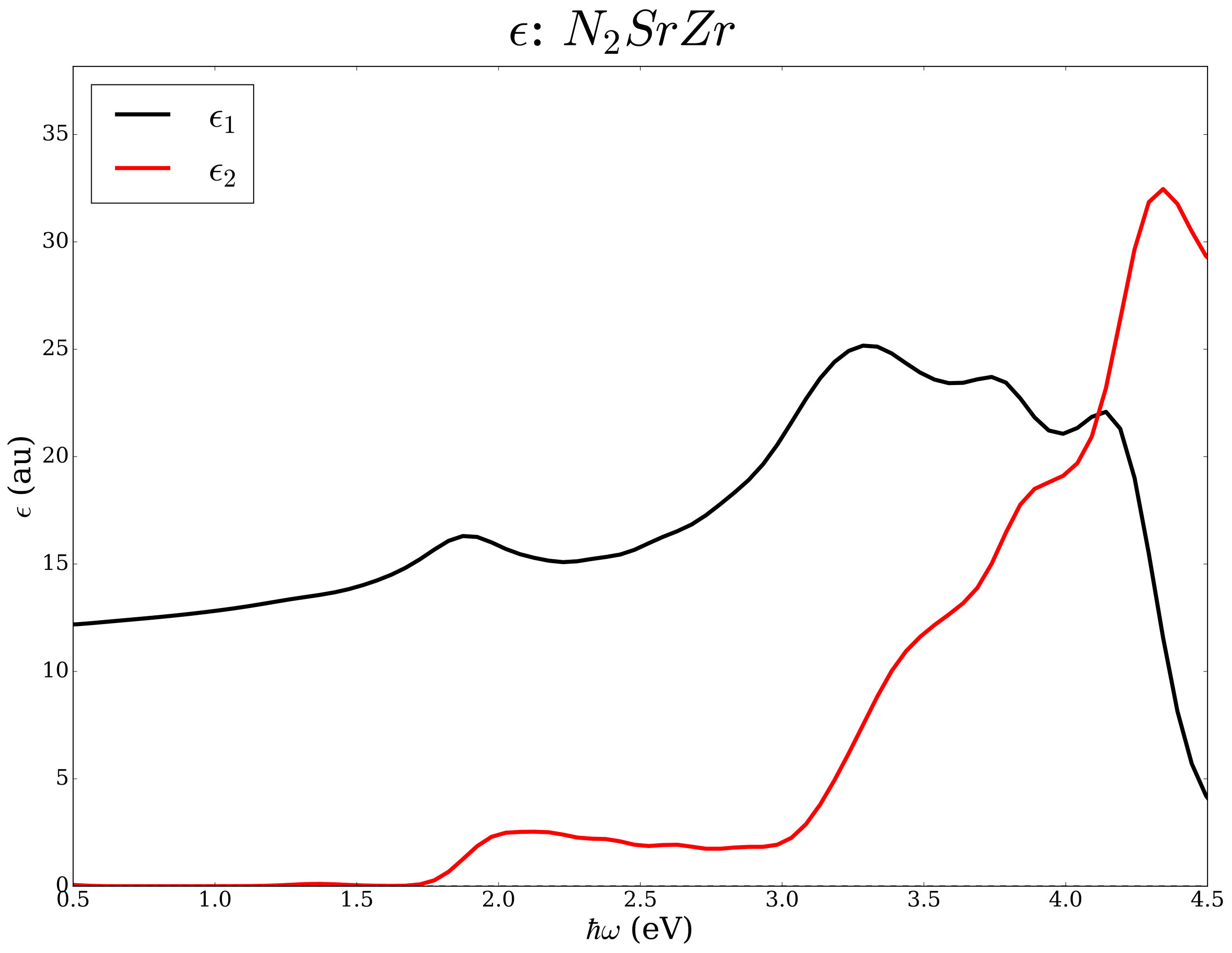 Figure 6
Figure 6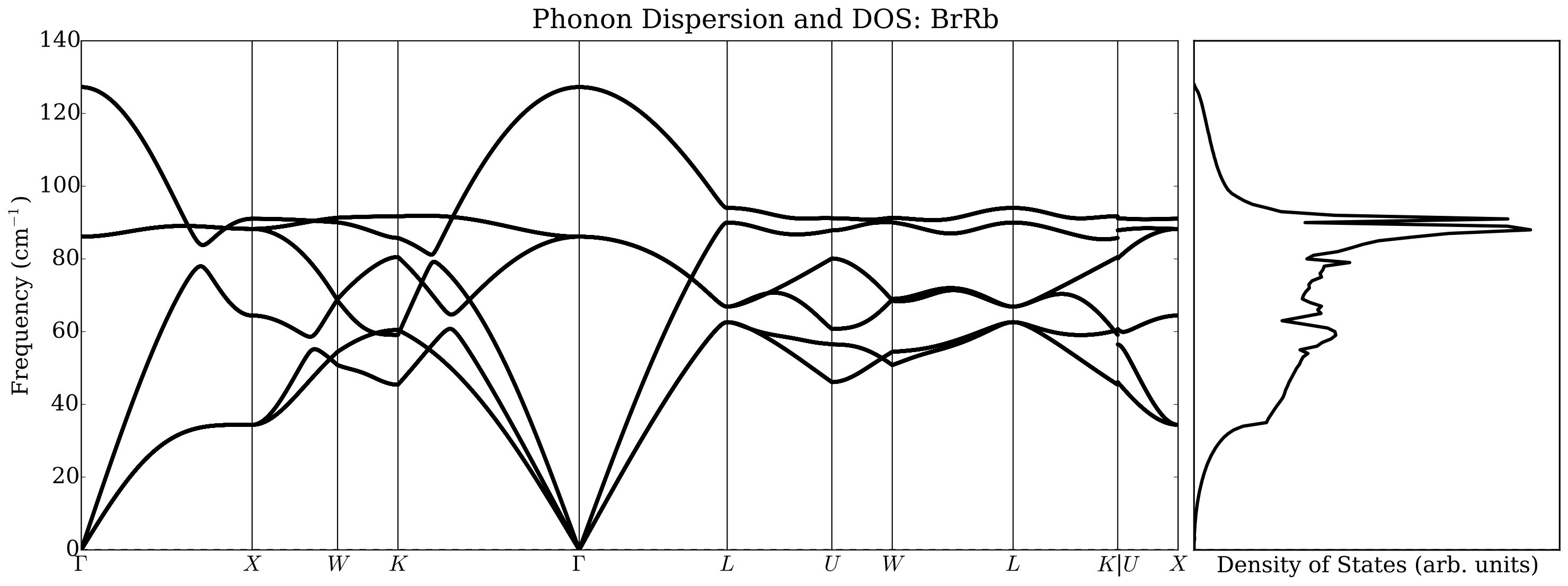 Figure 7
Figure 7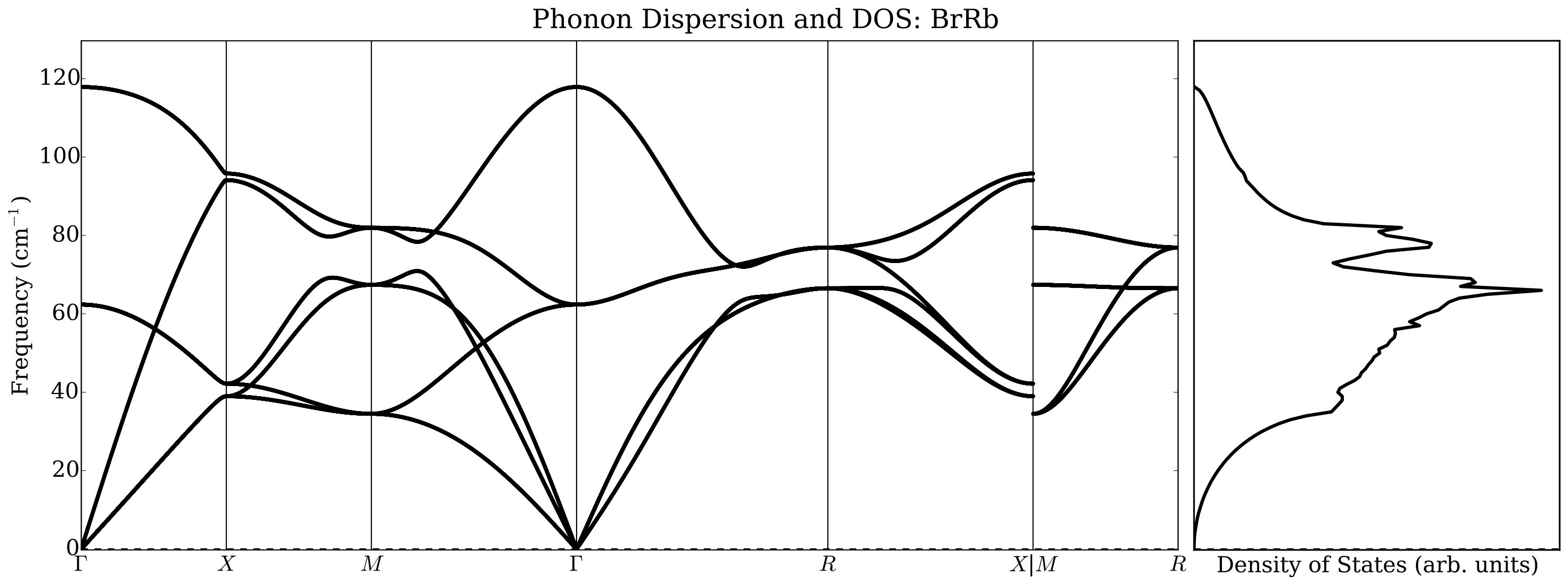 Figure 8
Figure 8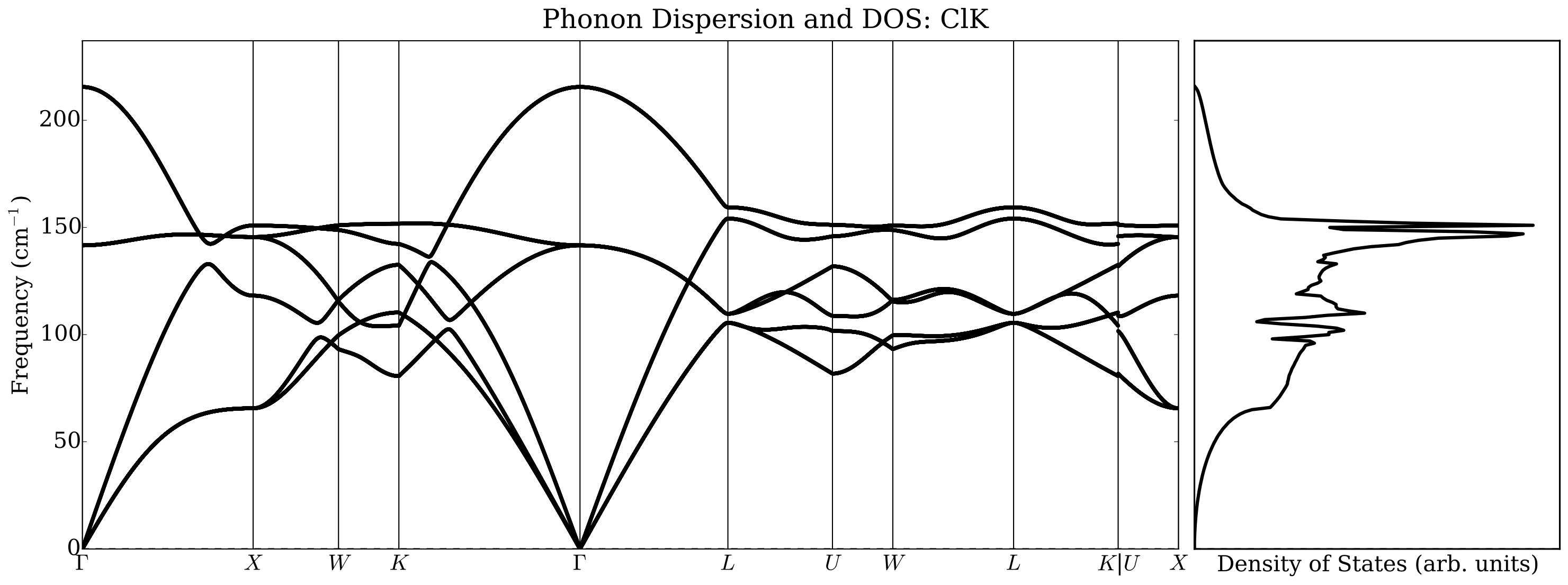 Figure 9
Figure 9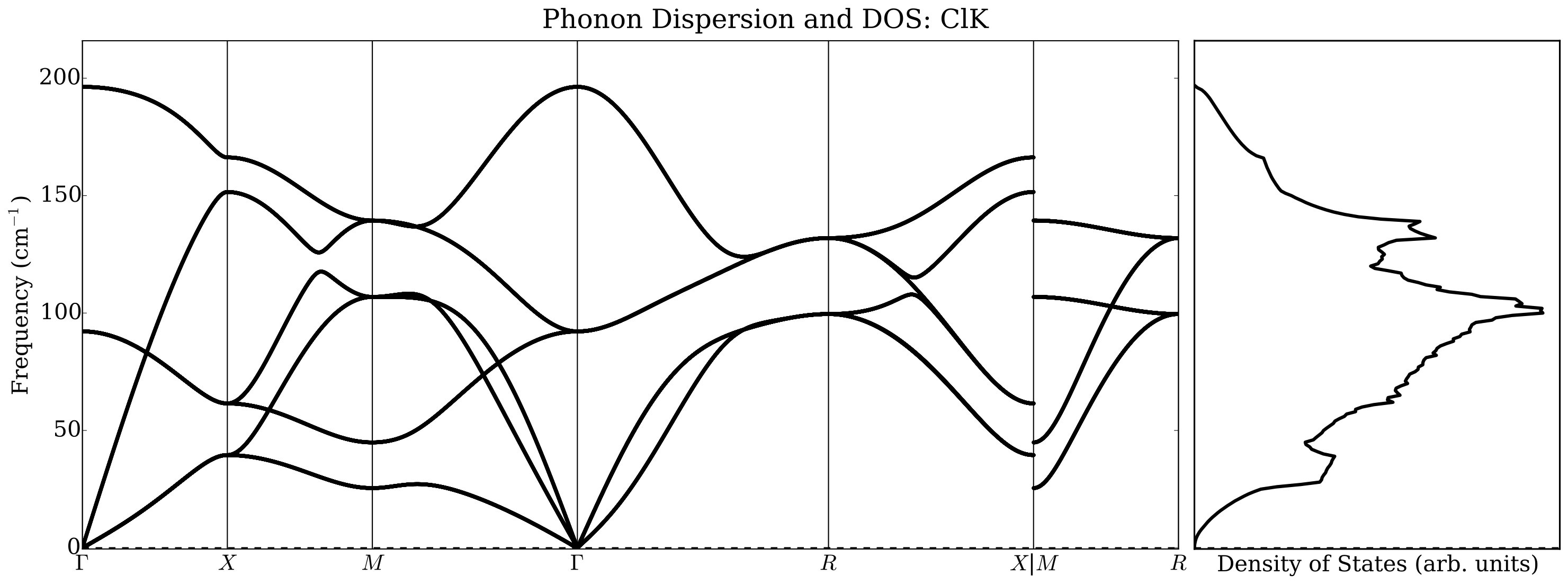 Figure 10
Figure 10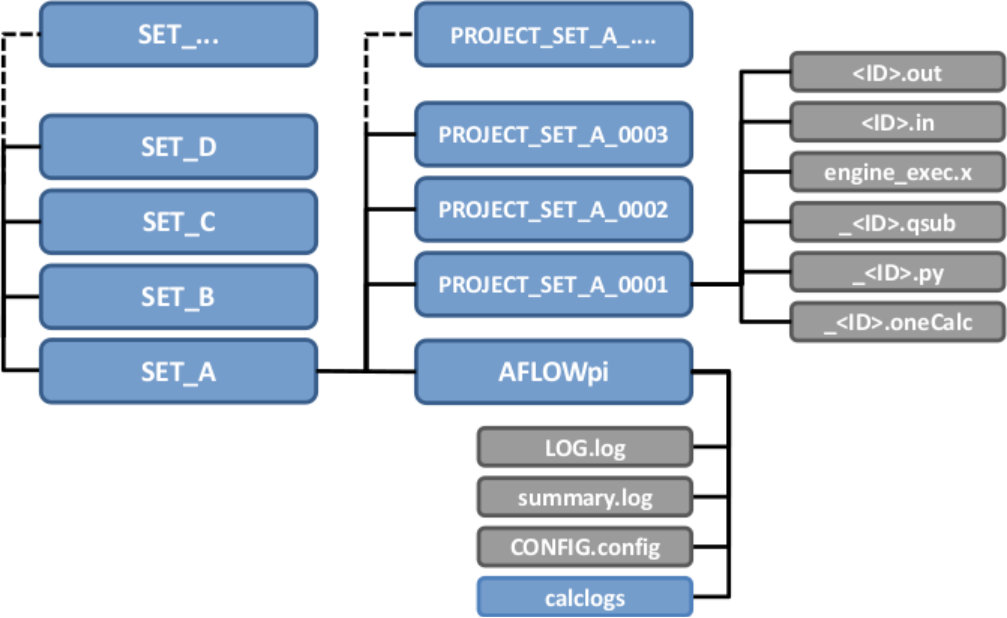 Figure 11
Figure 11| AlP | AlAs | AlSb | GaP | GaAs | InP | |
|---|---|---|---|---|---|---|
| 0.01 | 0.08 | 0.17 | 20.00 | 20.01 | 14.97 | |
| 2.31 | 2.12 | 1.11 | 1.87 | 1.75 | 0.00 | |
| 5.49 (5.46) | 5.74 (5.66) | 6.23 (6.13) | 5.53 (5.45) | 5.79 (6.05) | 5.97 (5.86) | |
| 5.44 (5.46) | 5.67(5.66) | 6.17(6.13) | 5.48(5.45) | 5.73(6.05) | 5.91 (5.86) | |
| 3.1 (3.6) | 1.8 (3.0) | 1.2 (2.3) | 1.3 (2.8) | 0.0 (0.8 ) | 0.4 (1.4) | |
| 3.9 (3.6) | 2.5 (3.0) | 1.7 (2.3) | 1.9 (2.8) | 0.5 (0.8) | 1.0 (1.4) | |
| 1.3 (2.5) | 1.5 (2.2) | 1.3 (1.7) | 2.1 (2.3) | 1.6 (1.1) | 1.8 (2.4) | |
| 2.1 (2.5) | 1.9 (2.2) | 1.5 (1.7) | 2.1 (2.3) | 1.9 (1.1) | 2.3 (2.4) | |
| 2.7 (2.5) | 1.7 (2.4) | 1.3 (2.3) | 1.4 (2.7) | 0.7 (0.9) | 1.1 (2.0) | |
| 3.3 (2.5) | 2.6 (2.4) | 1.5 (2.3) | 1.8 (2.7) | 1.1 (0.9) | 1.8 (2.0) |
| SrTiN2 | BaTiN2 | SrZrN2 | BaZrN2 | SrHfN2 | BaHfN2 | |
|---|---|---|---|---|---|---|
| U(A) | 0.04 | 3.69 | 0.04 | 4.32 | 0.05 | 4.40 |
| U(M) | 0.28 | 0.29 | 0.28 | 0.20 | 0.13 | 0.15 |
| U(N) | 3.09 | 2.93 | 3.09 | 2.80 | 3.06 | 2.80 |
| Eg | 1.25 | 1.02 | 1.85 | 1.58 | 2.21 | 1.87 |
| ( GPa) | ACBN0 | PBE | EXP | Model | |
|---|---|---|---|---|---|
| LiF | 113.6 | 111.5 | 113.9 | 104.6 | |
| 50.6 | 46.3 | 47.6 | 42.4 | ||
| 66.2 | 60.4 | 63.6 | 70.8 | ||
| LiCl | 51.5 | 52.1 | 60.7 | 66.6 | |
| 23.9 | 21.8 | 22.7 | 20.9 | ||
| 27.1 | 24.9 | 26.9 | 24.6 | ||
| NaF | 101.1 | 95.9 | 108.9 | 97.6 | |
| 24.8 | 22.2 | 22.9 | 22.9 | ||
| 29.4 | 26.4 | 28.9 | 29.9 | ||
| NaCl | 51.2 | 49.2 | 57.3 | 54.5 | |
| 13.4 | 12.0 | 11.2 | 11.2 | ||
| 13.9 | 12.3 | 13.3 | 11.6 | ||
| KF | 60.9 | 60.1 | 75.7 | 85.0 | |
| 14.9 | 14.1 | 13.5 | 14.7 | ||
| 14.5 | 13.6 | 13.3 | 17.1 | ||
| KCl | 37.6 | 36.2 | 40.9 | 53.8 | |
| 6.8 | 6.3 | 7.0 | 5.4 | ||
| 7.2 | 6.6 | 6.2 | 8.3 |
| LiF | LiCl | LiBr | NaF | NaCl | NaBr | KF | KCl | KBr | |
|---|---|---|---|---|---|---|---|---|---|
| U(A) | 0.14 | 0.04 | 0.01 | 0.19 | 0.02 | 0.02 | 0.17 | 0.03 | 0.02 |
| U(X) | 11.41 | 6.31 | 5.49 | 11.95 | 6.95 | 5.97 | 12.77 | 7.29 | 6.27 |
| aL | 3.99 | 5.06 | 5.41 | 4.60 | 5.57 | 5.89 | 5.32 | 6.24 | 6.55 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInorganic Chemistry and Materials · Machine Learning in Materials Science · Electronic and Structural Properties of Oxides
** AFLOW: A minimalist approach to high-throughput ab initio calculations including the generation of tight-binding hamiltonians**
Andrew R. Supka1,2, Troy E. Lyons1, Laalitha Liyanage3, Pino D’Amico4,5, Rabih Al Rahal Al Orabi1, Sharad Mahatara1, Priya Gopal1, Cormac Toher7,8, Davide Ceresoli6, Arrigo Calzolari3,4,5,8, Stefano Curtarolo3,8, Marco Buongiorno Nardelli2,8, and Marco Fornari1,2,8⋆
1 Department of Physics, Central Michigan University, Mount Pleasant MI, USA
2 Science of Advanced Materials Program, Central Michigan University, Mount Pleasant MI, USA
3Department of Physics and Department of Chemistry, University of North Texas, Denton TX, USA
4Dipartimento di Fisica, Informatica e Matematica, Universitá di Modena and Reggio Emilia, Via Campi 213/a, 41125 Modena, Italy
5CNR-NANO Research Center S3, Via Campi 213/a, 41125 Modena, Italy
6CNR-ISTM, Istituto di Scienze e Tecnologie Molecolari, I-20133 Milano, Italy
7Materials Science, Electrical Engineering, Physics and Chemistry, Duke University, Durham NC, 27708
8Center for Materials Genomics, Duke University, Durham, NC 27708, USA
*⋆*corresponding: [email protected]
Abstract
Tight-binding models provide a conceptually transparent and computationally efficient method to represent the electronic properties of materials. With AFLOW we introduce a framework for high-throughput first principles calculations that automatically generates tight-binding hamiltonians without any additional input. Several additional features are included in AFLOW with the intent to simplify the self-consistent calculation of Hubbard corrections, the calculations of phonon dispersions, elastic properties, complex dielectric constants, and electronic transport coefficients. As examples we show how to compute the optical properties of layered nitrides in the N2 family, and the elastic and vibrational properties of binary halides with CsCl and NaCl structure.
keywords:
High-throughput Calculations, Computer Simulations, Materials Databases
1 Introduction
Accelerated materials discovery is currently the focus of much infrastructure development both for experimental (see for instance Refs. [1, 2, 3, 4]) and theoretical research [5, 6, 7]. The key components always involve robust data generation or collection, real time feedback and error control, curation and archival of the data, and post-processing tools for analysis and visualization. The ultimate goal is to discover relationships that may be hidden in the large data sets and possibly predict materials with improved specific functionalities.
Flavors of density functional theory (DFT) usually form the basis for the generation of theoretical data since they provide reasonable transferability across different chemical compositions and structural themes at acceptable computational cost. Standard electronic structure packages such as Quantum Espresso [8] or VASP [9] are used as the engine of the calculations but an additional layer of software tools is needed to efficiently manage the high-throughput (HT) workflows. In addition, to streamline data generation, those software tools usually curate the data and prepare them for distribution in online electronic structure properties databases [10, 11, 12, 13] that can be mined a posteriori.
Several HT frameworks for electronic structure calculations were discussed in the literature. PYMATGEN [14] is the data generation software infrastructure behind the Materials Project database [10]. It is written in Python 2.7 using libraries such as Numpy and Scipy and exploits VASP and AbInit [15] as standard electronic structure engines. Many tools are available to study phase stability and phase diagrams. AiiDA [16] is an integrated software based on a paradigm involving automation, data, environment, and storage. It is written in Python 2.7 and revolves around relational databases for the overall design in addition to the storage component. For the automation component AiiDA involves workflows that use Quantum Espresso although other electronic structure codes are supported. In addition, scripting interfaces such ASE [17] can be used by AiiDA to control the workflow. ASE is paired with the Computational Materials Repository [18]. Additional HT software tools for density functional calculations are qmpy (associated with the Open Quantum Materials Database) [19] and the HT toolkit (HTTK associated with the Open Materials Database, http://httk.opendatabase.se); these software packages are written in Python and streamline electronic structure data generation. The framework AFLOW [20, 21, 22, 23, 24, 25, 26, 27] is an efficient tool for high-throughput calculations with VASP. It is written in C++ and includes tools to create input files for Quantum Espresso, to deal automatically with supercells and fractional occupations, to perform symmetry analysis, and to evaluate several thermodynamical and thermal transport properties at different levels of approximation [28, 29]. Data generated with AFLOW are available online on the AFLOW.org repositories [11, 30, 31] (see http://www.aflow.org).
With AFLOW we are complementing AFLOW with a minimalist software infrastructure that is easily portable, simple to use, and integrated with the AFLOW.org repositories. AFLOW has been initially developed for verification and testing purposes but has evolved into a modular software infrastucture that provides automatic workflows for the ab initio generation of tight-binding hamiltonians within the projected atomic orbital (PAO) scheme [32, 33, 34] and the self-consistent calculation of Hubbard corrections within the ACBN0 approach [35]. In addition, workflows for the calculation of phonon dispersions with Hubbard correction, the calculation of elastic constants, diffusive transport coefficients, and optical spectra are available. When possible, the code exploits the tight-binding hamiltonians as in Ref. [36].
2 Software design and data organization
AFLOW’s design provides a simple, task oriented Python package to construct large sets of calculations from one or few input files for a specific ab initio engine and then to perform a workflow of tasks for each calculation in the set. The infrastructure is modeled on a simple and common modus operandi that involves prototypical input files (reference files) that are modified for desired tasks. The chosen workflow instructs AFLOW how to modify the reference files and guides the process of submitting, running, and monitoring all calculations.
The software is implemented in Python 2.7 (using the Python standard library and Numpy, Scipy, CIFfile, and Matplotlib). The systematic use of regular expression (re module) to parse and modify the input and output files according the specific workflow makes the software very portable and expandable to a variety of electronic structure engines and tools. Currently, AFLOW encapsulates pw.x and several post-processing software from the Quantum Espresso package [8] as well as ElaStic [37]. In addition it uses features of findsym [38]. The generation and manipulation of the TB Hamiltonians is done with the engine PAO [39].
The calculations and the resulting data are organized according to the layer structure adopted by AFLOW [30]. The first layer in the directory hierarchy is the Project layer that can contain multiple instances of the next layer, the Set layer. The directories for the individual calculation pipelines in the set are named PROJECT_SET_XXXX with PROJECT being the project name, SET being the name of the set and XXXX being the index of the calculation (See Figure 1). Each calculation pipeline receives a unique identifier to keep track of each pipe of calculations during setup and runtime as well as for storage and retrieval of the results. The identifier is constructed as a CRC64 hash [40], which is a unique 16 character string representing the initial ab initio engine input file for each calculation pipeline. The input file is first cleaned of commented out content and unnecessary whitespace, then lines of text are sorted alphabetically. Cleaning and sorting is necessary to ensure that extra whitespace or the ordering of the parameters will not change the value of the hash. Files for each task, or step, in each calculation pipeline are designated by ID_XX where XX is the step number and ID is the identifier for that calculation pipeline. An input for the first step in a pipeline with the identifier 20b8c6f83a2ca9b would read c20b8c6f83a2ca9b_01.in. Files and directories within each calculation’s directory that are considered unneeded after runtime, often referred to as scratch, such as Python scripts generated by AFLOW, cluster submission files, and temporary files of the ab initio engine have a prepended underscore. For example, the filename of the Python executable for the second step in that pipeline would be _c20b8c6f83a2ca9b_02.py. AFLOW modularizes the calculation pipelines by separating each task into its own executable. When a task is completed, the next task is initiated and so on until the pipeline completes. The location of the directory for each calculation pipeline is preserved for all tasks the user issues for the set. All files needed to perform the tasks as well as all files generated by the tasks are stored in the folder pertaining to the individual calculation pipeline in order to ensure control and transferability.
Within each Set, AFLOW creates a directory named AFLOWpi that contains a master runtime log of the session, a copy of the configuration file used in the session, a summary log with relevant information from the output of every calculation within the set, as well as the loadable calculation files (LCFs). In order to ensure data provenance AFLOWpi includes mandatory information to be provided at the beginning of the data generation process. Each calculation pipeline has its own LCF that are pickled and used to control and monitor the workflow especially for resuming or expanding the calculations. AFLOW will run as a cluster job submission (currently PBS, http://pbsworks.com) or as a local process on systems without a cluster queue. The user has the freedom to choose how the steps in the workflows of the calculations are grouped in terms of cluster submissions.
Specific mechanisms for auto-restart and both manual and automatic error control have been implemented to manage the full set of calculations at once. The restart mechanism keeps track of the time a cluster job has been running and prepares calculations so that the electronic structure engine exits cleanly before the wall-time runs out. A wall-time buffer with a default of 10% of the total wall-time request allows time to save data to disk and, if local scratch option is used, for the temporary files in the local scratch diskspace to be copied to the global scratch. After cleaning up temporary files, the command to resubmit the cluster job is given and then the job exits. There are several locking mechanisms in place to ensure that there is no wasted calculation time. In addition to auto restarting, if there are major failures of the electronic structure engine, the user can load a calculation set and filter only the jobs where there were errors causing the execution pipeline to stop at a certain point and check what kind of errors occurred, if any. Calculations that failed can be filtered into a subset. Certain actions can be taken to help the subset to complete.
AFLOW’s source code includes documentation strings written in the Google Python docstring format for readability. These docstrings are located at the beginning of each function or class. AFLOW documentation is generated via the Sphinx Python package. Sphinx can be used to parse source code written in Python, C, C++ as well as several other programming languages to automatically generate user documentation from docstrings. We use Sphinx to generate LaTeX documentation for AFLOW as well as a searchable html documentation.
3 Generation of tight-binding hamiltonians
Theory and applications of the methodology used by AFLOW to generate tight-binding (TB) hamiltonians for first principles plane-wave pseudopotential calculations have been discussed in Refs. [32, 33, 34]. Our implementation does not require any additional input with respect to the electronic structure calculations and provides the real space representation of the TB matrix in self-contained XML format. The sparse TB matrix is represented on a grid as and can be easily Fourier transformed into
[TABLE]
and diagonalized to determine the full energy dispersions, , with the desired level of resolution in reciprocal space or to compute additional properties associated with derivatives in reciprocal space (linear momenta, and effective masses). The real space representation can also be used directly. We have recently applied these methodologies to study diffusive and ballistic transport as well as the optical properties in the independent particles approximation [36].
4 ACBN0 calculations
The TB representation of the electronic structure can be exploited to efficiently compute two-electron integrals for the development of local exchange functionals. AFLOW implements the direct and self-consistent evaluation of the on-site Hubbard correction that greatly improves the accuracy of standard DFT calculations [35]. Due to the accurate TB representation, the evaluation of the parameters for atoms in different chemical environments or close to topological defects becomes trivial and AFLOW facilitates the investigation of such systems. ACBN0 only demands computational resources comparable to regular LDA or PBE calculations. We have extensively investigated the improvements and the limits of ACBN0 calculation in Refs. [41, 42]. Overall, ACBN0 delivers better accuracy for lattice parameters and bulk moduli, improves the energy band gap on semiconductors and insulators as well as the relative position of occupied -manifolds in transition metal oxides, and optimizes the phonon dispersions and associated thermal transport properties.
5 Installation and data generation
The minimalist style of AFLOW makes it easy to install the software. One command to download the source code from its repository online, and two commands to enter the directory and to install AFLOW as an importable Python package. The setup.py in the base directory of the AFLOW source code tree is executed to build and make it importable in Python scripts. All the commands can be run interactively through any Python shell. Currently AFLOW includes three basic submodules: prep, run, scfuj to perform the basic calculations including ACBN0 and the generation of tight-binding hamiltonians. The regular expressions for interacting with a specific electronic structure engine (pw.x for instance) are stored separately. In addition, the plot, aflowlib, db, retr modules can be used to extract, post-process, and export the computed quantities. The distribution also includes the pseudo module for pseudo-potential testing. A detailed description of all the methods is included in the distribution as well as practical examples. A simple interface class allows for easy customization of calculation workflows using methods to instruct AFLOW to prepare the specific tasks.
The construction of a workflow starts with the command AFLOWpi.prep.init() which initiates the AFLOW session and requires a project name and the name of the configuration file as input. Specifying sets within a project is optional but highly recommended. AFLOWpi.prep.init() returns a session object which has methods to form the calculation sets. When one of the methods of the session object is called, it returns a “calculation_set” object. The calculation_set object contains methods to construct workflows which add tasks to be done for each calculation in the set. Let assume we plan to generate the PAO-TB hamiltonians for the III-V semiconductors (AlP, GaP, InP, AlAs, GaAs, AlSb, GaSb, and InSb) from the electronic structure generated without and with the ACBN0 approach. In a prototypical reference input file (e.g. mHT_TB.ref) the labels for the atomic species are substituted with the variables _AFLOWPI_A and _AFLOWPI_B. That input combined with the list of chemical substitutions is processed by AFLOW in order to prepare a pipeline of calculations starting with the determination of the ground state potential (session.scfs), the structural relaxations (calcs.vcrelax), and the generation of the TB hamiltonian (calcs.tight_binding) including plots for the densities of states and the band structures. Then the workflow continues with the self-consistent calculation of the Hubbard corrections (calcs.acbn0) before recalculating a new TB hamiltonian and new plots. At the end of the user’s AFLOW script, the command calcs.submit must be included to run the calculation workflow (see Figure 2). In Figs. 3 and 4, we show plots generated by AFLOW before and after the ACBN0 cycle. It is interesting to pinpoint the corrections of the band gap and the modification of the dispersion relationships (the Hubbard corrections determined running this workflow are in Table 1). All the tight-binding hamiltonians are stored as XML files and can be distributed independently. We stress the fact that the energy dispersion can be computed with ab initio accuracy.
Several styles may be used in AFLOW to define the calculations in a set. The first style is based on the cartesian product of two lists (as in Figure 2). The second style, labelled “zip mode”, generates all the entries by taking, in order, the corresponding entries in the keywords lists (see Section 7.2). In addition, calculation sets can be formed from a list of one or more input filenames for the electronic structure engine (these can be completed appropriately with the missing variables).
6 Data retrieval and exporting
AFLOW extracts and monitors data such as energy, structure, atomic positions, total force, total stress, and automatically records it in the summary.log file in the calculation set’s AFLOWpi directory. Quantities such as those listed above can be extracted for a given step of the calculation pipeline using the AFLOWpi.retr module.
In addition, results can be packaged for the online repository www.aflow.org by adding
set_name=’III-V’,
config=’./mHT_TB.config’)
to the workflow.
AFLOW also includes post-processing routines to compute and plot the radial distribution function, compare the energetics of different distortions, generate CIF files, and perform symmetry analysis.
7 Examples
7.1 Optical (and transport) properties
Layered nitrides with chemical formula N2 were studied earlier from the point of view of thermoelectric potentials and superconductivity [45, 46, 47, 48, 49, 50]. As an example we analyze the same chemical space (=Sr, Ba and M=Ti, Zr, Hf) from the point of view of the electronic transport in the constant relaxation time approximation and the optical properties within the independent electronic approximation. Using well converged basis sets and k-space sampling (see Ref. [45] for computational details), we have determined the electrical conductivity, the Seebeck coefficient, and the the complex dielectric functions with and without ACBN0. The workflow is in Figure 5.
We report data for N2 compounds in Table 2 and in Figure 6. Among the N2 compositions, the compounds containing Ti have absorption edges below the Shockley-Queisser limit of 1.34 eV; the compounds containing Hf and Zr exceed such a limit but seem potentially interesting as end points of (Ti,)N2 alloys with =Zr and Hf where tuning of the band gap can lead to improved photovoltaics potentials.
7.2 Phonons and elastic constants
In order to show how the calculations of phonon dispersions and of the elastic constants can be performed in AFLOW we analyzed a small chemical space of alkali halides with (=Li, Na, K and =F, Cl, Br) with the NaCl and the CsCl structures. The workflow is in Figure 7: notice the use of the zip mode to specify the chemical space and the use of two reference files for the NaCl and CsCl structure, respectively. Elastic constants are computed solely on the NaCl structure.
Under normal conditions, alkali halides have NaCl or B1-type crystal structure, but they transform under moderate pressure to the CsCl or B2-type structure. There has been much work done on thermal conductivity during the phase transition of alkali halides: Averkin [51] found that the phase transition completely eliminates the influence of the elastic anisotropy on the thermal conductivity, as well as its dependence on pressure. In that paper an empirical relation was also established between elastic anisotropy and thermal conductivity. However, Slack [52] concluded that there is no need to invoke the elastic anisotropy to explain thermal conductivity differences. He noticed that NaCl structure compounds have a higher thermal conductivity ratio than CsCl ones by about a factor of two and conjectured a large increase in the Grüneisen parameter as the co-ordination number changes from six to eight. Our results indicate, however, that a major contribution to the change in the thermal conductivity is related to the average sound velocity reduction in CsCl structure with respect to the NaCl structure. In Figure 8 we report, as an example, phonon dispersions automatically generated by AFLOW using a finite difference method. We did not observe significant changes using the ACBN0 approach with respect to the PBE calculations, and we choose to report selected ACBN0 data as an example.
The elastic constants of alkali halides are important as they are closely related to the ionic binding force in the crystal [44]. There are several phenomenological models to determine the elastic constants: in the central force model [53], the elastic constants are given by the sum of the terms due to long range and short range central forces. This model leads to a Cauchy relation , which is not true experimentally. The degree to which this Cauchy relation is not satisfied is a measure of the importance of many-body forces in lattice dynamics of the alkali halide which Lowdin [54] has found to exist. The breathing shell model [55] includes many-body forces and makes a prediction in alkali halides. Mahan [44] studied the effect of induced polarization on phonon modes at long wavelength and introduced a polarization term in the central force model predicting . Experimentally the deviation is found in both directions. We report in Table 3 the comparison between first principles calculations with and without ACBN0, experimental data, and results from Ref. [44]. Our results improve the agreement with experimental data with respect to the model in Ref. [44] and indicate the sensitivity of the elastic constants to the choice of the computational method.
8 Conclusions
We have presented here a minimalist high-throughput software infrastructure that, in addition to organizing and controlling the computational workflow, facilitates the generation of tight-binding Hamiltonians to represent accurately the electronic properties with ab initio accuracy. AFLOW exploits the tight-binding formalism to compute electronic transport and optical properties within the independent particle approximation. Three examples were used to illustrate the features of AFLOW (1) the calculation of the band structure of III-V semiconductors with and without the ACBN0 correction, (2) the study of the complex dielectric constant of N2 (=Sr, Ba and =Ti, Zr, Hf), and (3) the phonon dispersions and the elastic constants of binary halides ( with =Li, Na, K and =F, Cl, Br). We found that (Ti,)N2 alloys with =Zr and Hf may have potential for photovoltaic applications. In addition, we justified the dramatic changes in thermal conductivity occurring at the transition between NaCl and CsCl structure in terms of sound velocity. The code, including the examples presented in this paper, is available at www.aflow.org/aflowpi.
9 Acknowledgments
We are grateful to the High Performance Computing Center at Michigan State University and the Texas Advanced Computing Center at the University of Texas Austin. The members of the AFLOW Consortium (http://www.aflow.org) acknowledge support by DOD-ONR (N00014-13-1-0635, N00014-11-1-0136, N00014-15-1-2863). The authors also acknowledge Duke University — Center for Materials Genomics — and the CRAY corporation for computational support.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1[1] R. Potyrailo, K. Rajan, K. Stoewe, I. Takeuchi, B. Chisholm, and H. Lam, Combinatorial and High-Throughput Screening of Materials Libraries: Review of State of the Art , ACS Combinatorial Science 13 , 579–633 (2011).
- 2[2] K. Rajan, Combinatorial Materials Sciences: Experimental Strategies for Accelerated Knowledge Discovery , Annual Review of Materials Research 38 , 299–322 (2008).
- 3[3] I. Takeuchi, C. J. Long, O. O. Famodu, M. Murakami, J. Hattrick-Simpers, G. W. Rubloff, M. Stukowski, and K. Rajan, Data management and visualization of x-ray diffraction spectra from thin film ternary composition spreads , Review of Scientific Instruments 76 , 062223 (2005).
- 4[4] A. G. Kusne, T. Gao, A. Mehta, L. Ke, M. C. Nguyen, K.-M. Ho, V. Antropov, C.-Z. Wang, M. J. Kramer, C. Long, and I. Takeuchi, On-the-fly machine-learning for high-throughput experiments: search for rare-earth-free permanent magnets , Scientific Reports 4 , 6367 EP– (2014).
- 5[5] S. Curtarolo, G. L. W. Hart, M. Buongiorno Nardelli, N. Mingo, S. Sanvito, and O. Levy, The high-throughput highway to computational materials design , Nat. Mater. 12 , 191–201 (2013).
- 6[6] D. Morgan, G. Ceder, and S. Curtarolo, High-throughput and data mining with ab initio methods , Measurement Science and Technology 16 , 296 (2005).
- 7[7] S. Curtarolo, D. Morgan, and G. Ceder, Accuracy of ab initio methods in predicting the crystal structures of metals: A review of 80 binary alloys , Calphad 29 , 163–211 (2005).
- 8[8] P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari, and R. M. Wentzcovitch, QUANTUM ESPRESSO: a modular and open-source softwa